Namelists#
Initialization Parameters#
advanced_div_correction#
- Fortran Type: L
-
Default: .FALSE.
Parameter to enable an improved divergence correction method for the fifth-order scalar advection scheme.
advanced_div_correction = .T. activates a better divergence correction for scalar transports with the fifth-order advection scheme. This significantly improves the conservation of scalar quantities. The parameter becomes only effective if scalar_advec = 'ws-scheme' and requires loop_optimization = 'vector'.
The fifth-order Wicker-Skamarock scheme is only conditionally conservative, i.e. scalar conservation is achieved as long as the flow divergence is negligible. For topography = 'flat' this condition is usually guaranteed by the pressure solver. However, in case of topography /= 'flat' with vertical and elevated horizontal surfaces, the divergence that remains after the pressure solver has been called is not negligible any more at fluid grid points located in the vicinity of walls. There is a default removal of divergence errors implemented in the Wicker-Skamarock scheme, which works sufficiently well in most applications. However, for some cases this default correction is not sufficient and scalar is not fully conserved in the near wall regions, leading to changes in the domain mean concentration of a few percent over time. advanced_div_correction = .T. ensures for a much better conservation of scalar quantities.
Note:
Tests revealed that the Courant number should not exceed values >= 0.5. Therefore, it is recommended to set cfl_factor < 0.5. This correction method has still not thoroughly been tested yet for all kind of setups, and numerical instabilities may arise when using it. Please inform the PALM developers, if you experience such kind of issues.
allow_negative_scalar_values#
- Fortran Type: L
-
Default: .FALSE.
Parameter to steer the treatment of negative scalar values.
Due to numerical dispersion errors caused by non-monotone discretization of the advection term, numerical oscillations occur near strong gradients. This sometimes results in negative values of the scalar. By default, scalars are constrained to positive values (>= 0), i.e., in case a negative value appears after time stepping, the scalar value at a respective grid point is reset to 0.1 times the scalar value before the timestep. However, this way, the total scalar amount is not conserved any more and my increase during the simulation. With allow_negative_scalar_values = .T., negative scalar values are accepted and the total amount of the scalar in a source/sink-free domain is conserved during the simulation.
This option only becomes active for scalars that do not interact with the flow, i.e. for passive scalars (see passive_scalar), chemical species, and aerosols. Furthermore, also potential temperature, humidity, cloud-physical quantities, and the SGS-TKE are still constrained to positive values.
allow_roughness_limitation#
- Fortran Type: L
-
Default: .FALSE.
Parameter to define if automatic limitation of roughness lenghts shall be applied.
Application of Monin-Obukhov similarity requires the roughness length \(z_0\) to be smaller than the surface layer height \(z_{mo}\) (which is assumed as half the grid spacing in normal direction to the respective surface), because otherwise functions like \(ln \frac{z_{mo}}{z_0}\) give wrong results. It is automatically checked if roughness lengths for momentum, temperature, and mositure are \(\le 0.5 z_{mo}\) and the run is automatically aborted, if this condition is violated. Setting allow_roughness_limitation = .T. automatically limits the roughness lengths to \(0.5 z_{mo}\) and continues the run.
alpha_surface#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: degrees
Inclination of the model domain with respect to the horizontal.
A setting of alpha_surface /= 0.0 inclines the model domain along the x-direction with respect to the horizontal. This way, flows over inclined surfaces (e.g. drainage flows, gravity flows) can be simulated. In such a case, the buoyancy term appears in both, the equation of motion of the u-component and of the w-component.
An inclination is only possible in case of cyclic horizontal boundary conditions along
xandy(see bc_lr and bc_ns) and topography = 'flat'.For alpha_surface /= 0.0 the simultaneous use of humidity = .T. is not allowed.
Runs with inclined surface may require additional user-defined code as well as modifications to the default code. Please contact the PALM developer group in case you are planning to use this feature.
approximation#
- Fortran Type: C*20
-
Default: boussinesq
Parameter to choose the approximation of the model equations.
Currently 2 choices are available:
-
boussinesq
The Boussinesq approximation assumes incompressibility of the fluid. The density is assumed to be constant in space (horizontally and vertically) and in time. The constant density is calculated in accordance with the values given for the parameters surface_pressure and pt_surface.
Note: The specific value of the constant density only affects some of the diagnostic output quantities like the perturbation pressure or fluxes (see flux_output_mode). Prognostic quantities are not affected at all, i.e. their values are independent of the density.
-
anelastic
The anelastic approximation allows for a decrease of density with height. The density is still assumed to be constant along the horizontal directions and in time. The vertical profile of the density is computed based on the surface_pressure and the vertical profile of the potential temperature. The anelastic approximation requires momentum_advec = 'ws-scheme'. Furthermore, conserve_volume_flow = .T. is not supported.
-
bc_e_b#
- Fortran Type: C*20
-
Default: neumann
Bottom boundary condition of the subgrid-scale TKE.
The boundary condition is applied at all horizontal upward-facing walls accordingly. At vertical surfaces, e.g. building walls, a zero-gradient Neumann condition is always applied. At the top boundary a zero-gradient Neumann boundary condition is generally used, i.e.
e(nz+1) = e(nz).Currently 2 choices are available:
-
neumann
A zero-gradient Neumann is applied at horizontal surfaces, i.e.
e(k=0) = e(k=1), wheree(k=1)is computed via a the prognostic SGS-TKE equation. Elevated horizontal surfaces (e.g. rooftops) are treated the same. -
(u*)**2+neumann
For this case,
e(k=0) = e(k=1)is assumed too, but withe(k=1)= \((u_* / 0.1 )^2\), where \(u_*\) is the friction velocity, as calculated via Monin-Obukhov relations. Note, this alternative is just a test option and should be used with care.
-
bc_lr#
- Fortran Type: C*20
-
Default: cyclic
Boundary condition along
x(for all quantities).Remark concerning non-cyclic conditions:
In case of non-cyclic lateral boundaries, a Dirichlet condition is used at the inflow for all quantities, i.e. initial vertical profiles (see initializing_actions) are fixed during the run, except for the SGS-TKE, to which a zero-gradient Neumann condition is applied. At the outflow boundary, a radiation condition is used for all velocity components. A constant phase velocity is used, which is set to the maximum velocity allowed by the CFL criterion (i.e. for a Courant number of 1). For scalars, a Neumann condition is used. For the perturbation pressure, Neumann conditions are assumed both at the inflow and at the outflow. Further, for non-cyclic horizontal boundaries, call_psolver_at_all_substeps = .T. should be used.
In order to maintain a turbulent state of the flow in case of non-cyclic conditions, it may be necessary to continuously impose perturbations on the horizontal velocity field in the vicinity of the inflow throughout the entire simulation. One way to do this is to impose randomly distributed disturbances onto the inflow by using create_disturbances. The horizontal range to which these perturbations are applied is controlled by the parameters create_disturbances, inflow_disturbance_begin and inflow_disturbance_end. The vertical range and the perturbation amplitude are given by disturbance_level_b, disturbance_level_t, and disturbance_amplitude. The time interval at which perturbations are to be imposed is set by dt_disturb. Note that these inflow perturbances are added in addition to the standard perturbances that are activated with create_disturbances. The standard perturbances can be deactivated by setting the runtime-parameter disturbance_energy_limit = 0.0. A second approach to enable a continous turbulent inflow is to recycle the turbulent signal at a given recycling plane, or to read the turbulent inflow from the dynamic input file. For more information, please see turbulent_inflow_method and references therein.
Please note, usage of non-cyclic lateral boundaries requires very sensitive adjustments of the inflow (vertical profiles) and the bottom boundary conditions, e.g. a surface heating should may generate horizontal temperature gradients, inducing thermal circulations. Please check the model results very carefully. Detailed information can be found in the documentation of the non-cyclic lateral boundary conditions.
Todo: Treatment of bc_lr for offline nesting should be explained here.
Currently 4 choices are available:
-
cyclic
Periodic boundary conditions are assumed at the east and west domain boundary, i.e. the flow which leaves the model domain at the east domain boundary re-enters the domain at the west domain boundary.
In case of cyclic boundary conditions in both horizontal directions, a y-shift of boundary values at the left/right boundary can be introduced.
-
dirichlet/radiation
Inflow from left (west domain boundary) and outflow to right (east domain boundary). This requires the multigrid method to be used for solving the Poisson equation for perturbation pressure (see psolver), and it also requires cyclic boundary conditions along
y(see bc_ns). -
radiation/dirichlet
Inflow from right (east domain boundary) and outflow to left (west domain boundary). This requires the multigrid method to be used for solving the Poisson equation for perturbation pressure (see psolver) and it also requires cyclic boundary conditions along
y(see bc_ns). -
nested
This setting shouldn't be required, because in a 3d-nested run, the child domain boundary condition is automatically set to 'nested' by default, except for nesting_bounds = 'vertical_only' or 'cyclic_along_y'. In the latter cases bc_lr can be explicitely set to any allowed value /= 'nested'.
-
bc_ns#
- Fortran Type: C*20
-
Default: cyclic
Boundary condition along
y(for all quantities).Please see bc_lr for remarks concerning non-cyclic boundary conditions.
Currently 4 choices are available:
-
cyclic
Periodic boundary conditions are assumed at the north and south domain boundary, i.e. the flow which leaves the model domain at the north domain boundary re-enters the domain at the south domain boundary.
-
dirichlet/radiation
Inflow from north and outflow to south. This requires the multigrid method to be used for solving the Poisson equation for perturbation pressure (see psolver), and it also requires cyclic boundary conditions along
x(see bc_lr). -
radiation/dirichlet
Inflow from south and outflow to north. This requires the multigrid method to be used for solving the Poisson equation for perturbation pressure (see psolver) and it also requires cyclic boundary conditions along
x(see bc_lr). -
nested
This setting shouldn't be required, because in a 3d-nested run, the child domain boundary condition is automatically set to 'nested' by default, except for nesting_bounds = 'vertical_only' or 'cyclic_along_x'. In the latter cases bc_ns can be explicitely set to any allowed value /= 'nested'.
-
bc_p_b#
- Fortran Type: C*20
-
Default: neumann
Bottom boundary condition of the perturbation pressure.
Depending on the given value, the boundary condition is applied at the model domain bottom, i.e. at grid level
k=0. Since vertical velocity is zero at the rigid lower boundary (w(k=0) = 0.0), the consistent Neumann condition bc_p_b = 'neumann' (i.e. \(\frac{\partial p}{\partial z} = 0\)) should be used, which leaves the vertical componentwunchanged at this level, when the pressure solver is applied. However, at elevated physical surfaces, i.e. terrain or buildings, no pressure boundary condition is applied at all. Simultaneous use of the Neumann boundary conditions both at the bottom and at the top boundary (bc_p_t) is allowed and recommended.Note: In case of simultaneous use of Neumann boundary conditions both at the top and bottom, the the multigrid solver (see psolver = 'multigrid') calculates the perturbation pressure except for a (non-determined) constant offset. The global average value of perturbation pressure remains arbitrary. The convergence rate of the multigrid solver is usually lower than for mixed Dirichlet/Neumann conditions. Proper adjustment of steering parameters for the multigrid method may help to improve the convergence. In case of output of pressure perturbation data, you may remove the global offset manually.
Currently 2 choices are available:
-
dirichlet
A Dirichlet condition is applied with
p(k=0) = 0.0. -
neumann
A zero-gradient Neumann is applied at the model bottom, i.e.
p(k=0) = p(k=1).
-
bc_p_t#
- Fortran Type: C*20
-
Default: neumann
Top boundary condition of the perturbation pressure.
Depending on the given value, the boundary condition is applied at the model domain top, i.e. at
k=nz+1. For more information on pressure boundary conditions see bc_p_b.In case that the multigrid solver is used (see psolver), the default value is set to 'dirichlet'.
In case of nested runs, the default value of bc_p_t in the nest domains is not 'dirichlet' but 'neumann' instead, which will be set automatically. For the root domain of a nested run the default is 'dirichlet' as usual. For child domains see the note that is given for bc_p_b.
Currently 2 choices are available:
-
dirichlet
A Dirichlet condition is applied with
p(k=nz+1) = 0.0. -
neumann
A zero-gradient Neumann is applied at the model top, i.e.
p(k=nz+1) = p(k=nz).
-
bc_pt_b#
- Fortran Type: C*20
-
Default: dirichlet
Bottom boundary condition of the potential temperature at physical surfaces.
In the atmosphere part of coupled ocean-atmosphere runs, bc_pt_b is internally set and does not need to be prescribed.
bc_pt_b is allowed to be set as runtime parameter as well. In case of a restart run, it then overwrites the setting from the previous run and the new value will be used for further restarts, as long as no new setting via the runtime parameter namelist is made.
Currently 2 choices are available:
-
dirichlet
This case,
pt(k=0) = const =pt_surface + pt_surface_initial_change + pt_surface_heating_rate * time_since_3d_model_start. The value may be changed during the run using user-defined code. The resulting surface heat flux is then calculated via flux-gradient relationships using Monin-Obukhov similarity theory.If the radiation-module is switched on, surface temperatures are internally calculated by solving the surface energy balance equation.
-
neumann
A zero-gradient Neumann condition is applied at physical surfaces, i.e.
pt(k) = pt(k+1). A Neumann boundary condition is also used at vertical walls, e.g. building facades. A Neumann condition must be used when a surface_heatflux is prescribed, because otherwise the resolved scale may contribute to the surface flux so that the constant value as given by surface_heatflux would not exactly be guaranteed.
-
bc_pt_t#
- Fortran Type: C*20
-
Default: initial_gradient
Top boundary condition of the potential temperature at grid point
k=nzt+1.Note that due to the staggered grid the prognostic equation for temperature is solved up to (including)
k=nz.In case of nested runs, the default value of bc_pt_t in the child domains is 'nested' instead of 'initial_gradient'.
Currently 4 choices are available:
-
dirichlet
This case,
pt(k=nz+1) =const. The value is given by the initial temperature profile. This value does not change during the simulation. -
initial_gradient
The value of the temperature gradient at the top is calculated from the initial temperature profile (see pt_surface, pt_vertical_gradient) by
initial_gradient = ( pt_init(k=nz+1) - pt_init(k=nz) ) / dzu(nz+1). Using this value (assumed constant during the simulation), the temperature boundary values are calculated aspt(k=nz+1) = pt(k=nz) + initial_gradient * dzu(nz+1). -
nested
In child domains a Dirichlet boundary condition is automatically assumed. Boundary values are obtained from the respective parent domain. Explicitly setting bc_pt_t ='nested' is not required.
-
neumann
A zero-gradient Neumann condition is applied at the domain top, i.e.
pt(k=nz+1) = pt(k=nz). A Neumann condition must be used when a top_heatflux is prescribed, because otherwise the resolved scale may contribute to the top flux so that the constant value as given by top_heatflux would not exactly be guaranteed.
-
bc_q_b#
- Fortran Type: C*20
-
Default: dirichlet
Bottom boundary condition of the water vapor / total mixing ratio at physical surfaces.
Currently 2 choices are available:
-
dirichlet
This case,
q(k=0) = const =q_surface + q_surface_initial_change. The value may be changed during the run using user-defined code. The resulting surface latent heat flux is then calculated via flux-gradient relationships using Monin-Obukhov similarity theory. -
neumann
A zero-gradient Neumann condition is applied at physical surfaces, i.e.
q(k) = q(k+1). A Neumann condition must be used when a surface_waterflux is prescribed, because otherwise the resolved scale may contribute to the surface flux so that the constant value as given by surface_waterflux would not exactly be guaranteed.
-
bc_q_t#
- Fortran Type: C*20
-
Default: neumann
Top boundary condition of the water vapor / total water mixing ratio.
Note that due to the staggered grid the prognostic equation for the mixing ratio is solved up to (including)
k=nz.In case of nested runs, the default value of bc_q_t in the child domains is 'nested' instead of 'neumann'.
Currently 4 choices are available:
-
dirichlet
This case,
q(k=nz+1) =const. The value is given by the initial humidity profile. This value does not change during the simulation. -
initial_gradient
A Neumann condition is applied at the model top. The value of the humidity gradient at the top is calculated from the initial humidity profile (see q_surface, q_vertical_gradient) by
initial_gradient = ( q_init(k=nz+1) - q_init(k=nz) ) / dzu(nz+1). Using this value (assumed constant during the simulation), the humidity boundary values are calculated asq(k=nz+1) = q(k=nz) + initial_gradient * dzu(nz+1). -
nested
In child domains a Dirichlet boundary condition is automatically assumed. Boundary values are obtained from the respective parent domain. Explicitly setting bc_q_t = 'nested' is not required.
-
neumann
A zero-gradient Neumann condition is applied at the model top, i.e.
q(nz+1) = q(nz).
-
bc_s_b#
- Fortran Type: C*20
-
Default: dirichlet
Bottom boundary condition of the scalar concentration.
Currently 2 choices are available:
-
dirichlet
This case,
s(k=0) = const =s_surface + s_surface_initial_change. The value may be changed during the run using user-defined code. The resulting surface scalar flux is then calculated via flux-gradient relationships using Monin-Obukhov similarity theory. -
neumann
A zero-gradient Neumann condition is applied at physical surfaces, i.e.
s(k) = s(k+1). A Neumann condition must be used when a surface_scalarflux is prescribed, because otherwise the resolved scale may contribute to the surface flux so that the constant value as given by surface_scalarflux would not exactly be guaranteed.
-
bc_s_t#
- Fortran Type: C*20
-
Default: initial_gradient
Top boundary condition of the scalar concentration.
Note that due to the staggered grid the prognostic equation for the scalar is solved up to (including)
k=nz.In case of nested runs, the default value of bc_s_t in the child domains is 'nested' instead of 'initial_gradient'.
Currently 4 choices are available:
-
dirichlet
This case,
s(k=nz+1) =const. The value is given by the initial scalar profile. This value does not change during the simulation. -
initial_gradient
The value of the passive_scalar gradient at the top is calculated from the initial scalar concentration profile (see s_surface, s_vertical_gradient) by
initial_gradient = ( s_init(k=nz+1) - s_init(k=nz) ) / dzu(nz+1). Using this value (assumed constant during the simulation), the scalar boundary values are calculated ass(k=nz+1) = s(k=nz) + initial_gradient * dzu(nz+1). -
nested
In child domains a Dirichlet boundary condition is automatically assumed. Boundary values are obtained from the respective parent domain. Explicitly setting bc_s_t = 'nested' is not required.
-
neumann
A zero-gradient Neumann condition is applied at the model top, i.e.
s(k=nz+1) = s(k=nz).
-
bc_uv_b#
- Fortran Type: C*20
-
Default: dirichlet
Bottom boundary condition of the horizontal velocity components
uandv.Currently 2 choices are available:
-
dirichlet
No-slip condition with
u = v = 0.0at the bottom physical surface. -
neumann
Free-slip condition with
u(k=0) = u(k=1)andv(k=0) = v(k=1). The free-slip condition is not alloed with a constant flux layer switched on (see constant_flux_layer) at the bottom boundary.Note: Free-slip conditions are not realized for vertical walls.
-
bc_uv_t#
- Fortran Type: C*20
-
Default: dirichlet
Top boundary condition of the horizontal velocity components
uandv.Note that due to the staggered grid the prognostic equation for the horizontal velocity components are solved up to (including)
k=nz.For coupled ocean-atmosphere runs, bc_uv_t is internally set to 'neumann' and does not need to be prescribed.
Currently 4 choices are available:
-
dirichlet
This case, the top boundary values are set to the respective geostrophic wind values, i.e.
u(k=nz+1) = ug(nz+1)andv(k=nz+1) = vg(nz+1). Values do not change during the simulation. See ug_surface and vg_surface on how to set the vertical profile of the geostrophic wind. -
dirichlet_0
This special condition can be used for channel flows. It yields the no-slip condition
u(k=nz+1) = ug(nz+1) = 0.0andv(k=nz+1) = vg(nz+1) = 0.0. -
nested
In child domains a Dirichlet boundary condition is automatically assumed. Boundary values are obtained from the respective parent domain. Explicitly setting bc_uv_t = 'nested' is not required.
-
neumann
Free-slip conditions with
u(k=nz+1) = u(k=nz)andv(k=nz+1) = v(k=nz).
-
building_height#
- Fortran Type: R
- Default: 50.0
-
SI-Unit: m
Height of a single building.
building_height must be less than the height of the model domain. This parameter only becomes active if topography = 'single_building'.
building_length_x#
- Fortran Type: R
- Default: 50.0
-
SI-Unit: m
Width of a single building along x-direction.
building_length_x must be at least 3 * dx and no more than ( nx - 1 ) * dx - building_wall_left. This parameter only becomes active if topography = 'single_building'.
building_length_y#
- Fortran Type: R
- Default: 50.0
-
SI-Unit: m
Width of a single building along y-direction.
building_length_y must be at least 3 * dy and no more than ( ny - 1 ) * dy - building_wall_south. This parameter only becomes active if topography = 'single_building'.
building_wall_left#
- Fortran Type: R
- Default: Depends on nx
-
SI-Unit: m
x-coordinate of the left building wall (distance between the left building wall and the left border of the model domain).
building_wall_left must be at least 1 * dx and less than ( nx - 1 ) * dx - building_length_x. This parameter only becomes active if topography = 'single_building'.
By default, the building is centered along x-direction, i.e. building_wall_left = ( ( nx + 1 ) * dx - building_length_x ) / 2. Due to the staggered grid, the building will be displaced by -0.5 dx along x-direction and -0.5 dy along y-direction.
building_wall_south#
- Fortran Type: R
- Default: Depends on ny
-
SI-Unit: m
y-coordinate of the south building wall (distance between the south building wall and the south border of the model domain).
building_wall_south must be at least 1 * dy and less than ( ny - 1 ) * dy - building_length_y. This parameter only becomes active if topography = 'single_building'.
By default, the building is centered along y-direction, i.e. building_wall_south = ( ( ny + 1 ) * dy - building_length_y ) / 2. Due to the staggered grid, the building will be displaced by -0.5 dx along x-direction and -0.5 dy along y-direction.
calc_soil_moisture_during_spinup#
- Fortran Type: L
-
Default: .FALSE.
Parameter for switching on the water transport equation in the soil model during the spinup phase (see spinup_time).
If set to .T., an additional prognostic equation for the volumetric moisture content of the soil layer is solved.
call_psolver_at_all_substeps#
- Fortran Type: L
-
Default: .TRUE.
Switch to steer the call of the pressure solver.
By default, the Poisson equation for perturbation pressure (see psolver) is solved after each Runge-Kutta sub-timestep (see timestep_scheme). In order to speed-up performance, call_psolver_at_all_substeps = .F. forces the pressure solver to be called only at the last substep of the Runge-Kutta schemes. In some cases this already may sufficiently reduce the divergence of the velocity field. Nevertheless, small-scale ripples with wavelength \(2 \Delta\) may be seen in the results.
Note: The pressure solver must be called at each Runge-Kutta substep when momentum_advec = 'ws-scheme'.
Warning: Setting call_psolver_at_all_substeps = .F. may force the simulation to become unstable. Please carefully check the run-control file for if the divergence is sufficiently reduced.
canyon_height#
- Fortran Type: R
- Default: 50.0
-
SI-Unit: m
Street canyon height.
canyon_height must be less than the height of the model domain. This parameter only becomes active if topography = 'single_street_canyon'.
canyon_wall_left#
- Fortran Type: R
- Default: Depends on nx
-
SI-Unit: m
x-coordinate of the left canyon wall (distance between the left canyon wall and the left border of the model domain).
canyon_wall_left must be at least 1 * dx and less than ( nx - 1 ) * dx - canyon_width_x. This parameter only becomes active if topography = 'single_street_canyon'.
By default, the canyon is centered along x-direction, i.e. canyon_wall_left = ( ( nx + 1 ) * dx - canyon_width_x ) / 2.
canyon_wall_south#
- Fortran Type: R
- Default: Depends on ny
-
SI-Unit: m
y-coordinate of the south canyon wall (distance between the south canyon wall and the south border of the model domain).
canyon_wall_south must be at least 1 * dy and less than ( ny - 1 ) * dy - canyon_width_y. This parameter only becomes active if topography = 'single_street_canyon'.
By default, the canyon is centered along y-direction, i.e. building_wall_south = ( ( ny + 1 ) * dy - canyon_width_y ) / 2.
canyon_width_x#
- Fortran Type: R
- Default: undefined
-
SI-Unit: m
Width of a single street canyon along x-direction.
canyon_width_x must be at least 3 * dx and no more than ( nx - 1 ) * dx - canyon_wall_left. This parameter only becomes active if topography = 'single_street_canyon'. No value given implies a canyon orientation along y-direction and respective setting of canyon_width_y.
canyon_width_y#
- Fortran Type: R
- Default: undefined
-
SI-Unit: m
Width of a single street canyon along y-direction.
canyon_width_y must be at least 3 * dy and no more than ( ny - 1 ) * dy - canyon_wall_south. This parameter only becomes active if topography = 'single_street_canyon'. No value given implies a canyon orientation along x-direction and respective setting of canyon_width_x.
cfl_factor#
- Fortran Type: R
-
Default: Depends on timestep_scheme
Factor limiting the time step as given by the Courant-Friedrichs-Levy (CFL) and other criteria.
The internal model timestep
dt_3daccording to the CFL- and diffusion-criterion (as well as other criteria) is reduced bydt_3d* cfl_factor in order to avoid stability problems which may arise in the vicinity of the maximum allowed time step. The condition 0.0 < cfl_factor < 1.0 applies.The default value of cfl_factor depends on the timestep_scheme used: For the third order Runge-Kutta scheme (timestep_scheme = 'runge-kutta-3') it is cfl_factor = 0.9, for the second-order Runge-Kutta scheme (timestep_scheme = 'runge-kutta-2') a value of 0.8 is used, and for the Euler scheme (timestep_scheme = 'euler') cfl_factor = 0.1 applies.
Attention:
The cfl_factor does not imply if a fixed time step has been set via dt.
check_realistic_q#
- Fortran Type: L
-
Default: .TRUE.
Parameter to switch on/off a check if simulation is allowed to start under supersaturated conditions.
For check_realistic_q = .T., the run is stopped right at the beginning if the mixing ratio exceeds the saturation mixing ratio (as determined from the initial temperature profile) by more than 2%.
cloud_droplets#
- Fortran Type: L
-
Default: .FALSE.
Parameter to switch on the Lagrangian cloud model (LCM).
For cloud_droplets = .T. the Lagrangian particle model is used as a cloud model. Therefore, particles represent droplets and aerosols. Since it is computationally not feasible to simulate a realistic amount of droplets/particles, a single Lagrangian particle represents an ensemble of identical particles (with same radius, velocity, mass of solute aerosol, etc.) and is referred to as a "super-droplet". The number of droplets/particles in this ensemble is referred to as the "weighting factor" (see initial_weighting_factor. The LCM must be steered via namelist particle_parameters.
collective_wait#
- Fortran Type: L
-
Default: .FALSE.
Set barriers in front of collective MPI operations.
Via collective_wait = .T. barriers (
MPI_BARRIER) will be set in front of collective MPI operations (e.g.MPI_ALLTOALL,MPI_ALLREDUCE). Depending on the communication network in use, this may speed up the MPI data transfer and hence the performance of the code. So far, speed-up has only been observed for fat-tree networks.
conserve_volume_flow#
- Fortran Type: L
-
Default: .FALSE.
Conservation of volume flow along
x- andy-direction.In case of lateral cyclic boundary conditions (bc_lr = 'cyclic' and bc_ns = 'cyclic'), conserve_volume_flow = .T. guarantees that the volume flow through the xz- and yz-cross-sections of the total model domain remains constant at each time step throughout the simulation depending on the chosen conserve_volume_flow_mode.
In case of non-cyclic lateral boundary conditions (bc_lr /= 'cyclic' and bc_ns /= 'cyclic') conserve_volume_flow = .T. guarantees that the volume flow at the outflow boundary equals at each time step the volume flow at the inflow.
Volume flow conservation is obtained via adding or subtracting a constant value to/from the respective velocity components. For cyclic boundary conditions velocities at all grid points are adjusted with the same offset, for non-cyclic conditions only wall normal velocity components at the outflow are adjusted.
Note that conserve_volume_flow = .T. requires dp_external = .F..
In case of non-cyclic lateral boundary conditions, more detailed information about the conservation of volume flow is given in the documentation.
Warning:
Volume flow conservation acts non-physically on the flow physics! This might end up in wrong vertical profiles of horizontal velocities. E.g., a main run with topography/buildings that follows a 'flat' prerun will show an increase in horizontal velocities above the topography, because there is no volume flow below the topography any more, and since the initial velocity profiles used for determining the required volume flow are those of the prerun.
conserve_volume_flow_mode#
- Fortran Type: C*16
-
Default: initial_profiles
Parameter to set which velocities define the target volume flow.
conserve_volume_flow_mode only comes into effect if conserve_volume_flow = .T..
Currently 2 choices are available:
-
initial_profiles
The target volume flow is calculated at
t=0from the initial profiles ofuandv. This option is also standard for non-cyclic lateral boundary conditions (bc_lr /= 'cyclic' or bc_ns /= 'cyclic') because the (spanwise averaged) inflow profiles do not vary with time and are identical with the initial profiles.In case of a main run that follows a prerun the initial profiles are given by / taken from the prerun.
-
bulk_velocity
The target volume flow is calculated from predefined bulk velocities (see u_bulk and v_bulk). This setting is only allowed for cyclic lateral boundary conditions.
-
constant_flux_layer#
- Fortran Type: L
-
Default: .TRUE.
Parameter to switch on a constant flux layer at physical surfaces.
By default, a constant flux layer is switched on at the bottom boundary between z = 0 and z = 0.5 * dz (or in case of terrain, buildings, etc., the first computational grid point above ground for
u,vand the scalar quantities). For this setting, free-slip conditions foruandv(see bc_uv_b) are not allowed at the bottom boundary. Likewise, setting a constant eddy diffusivity (km_constant) like it is done in DNS mode does not allow constant_flux_layer = .T.. Within the constant flux layer Monin-Obukhov similarity is used to calculate the turbulent vertical fluxes based on stability, roughness length, and values of the quantities (velocity, temperature, etc.) at the bottom and top of the constant flux layer.The roughness length can be declared via parameters roughness_length and z0h_factor.
In case that a constant flux layer is explicitly switched off via constant_flux_layer = .F., but surface fluxes shall be prescribed (e.g. by setting surface_heatflux), it is required to set the parameter use_surface_fluxes = .T..
coupling_start_time#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: s
Simulated time of a precursor run.
Sets the time period a precursor run shall run uncoupled. This parameter is used to set up the precursor run control for atmosphere-ocean-coupled runs. It has to be set individually to the atmospheric / oceanic precursor run. The time in the data output will show negative values during the precursor run. See coupled runs for further information.
cut_cell_topography#
- Fortran Type: L
-
Default: .FALSE.
Parameter to control topography representation.
By default, PALM represents topography on a Cartesian grid, where a grid cell is either fully fluid or fully topography. By setting cut_cell_topography = .T., topography is represented with a cut-cell approach, where a grid cell can be partly fluid and partly obstacle, and where the orientation of the topography surface is not bound to the Cartesian directions any more.
Note:
A mixture of Cartesian step-like surfaces and cut-cell surfaces is currently not possible, e.g. vertically oriented building walls can not be represented via cut-cell surfaces.
cycle_mg#
- Fortran Type: C*1
-
Default: w
Type of cycle to be used with the multigrid method.
This parameter determines which type of cycle is applied in the multigrid method used for solving the Poisson equation for perturbation pressure (see psolver). It defines in which way it is switched between the fine and coarse grids. So-called v- and w-cycles are realized. The computational cost of w-cycles is much higher than that of v-cycles, however, w-cycles give a much better convergence, so using w-cycles is recommended in general.
Currently 2 choices are available:
-
v
v-cycle. The computational cost of v-cycles is significantly lower compared to w-cycles, though it gives a poorer convergence.
-
w
w-cycle. The computational cost of w-cycles is significantly higher compared to v-cycles, though it gives a better convergence.
-
damp_level_1d#
- Fortran Type: R
- Default: Value of zu(nz+1)
-
SI-Unit: m
Height where the damping layer begins in the 1d-model.
This parameter is used to switch on a damping layer for the 1d-model, which is generally needed for the damping of inertial oscillations. Damping is done by gradually increasing the value of the eddy diffusivities about 10% per vertical grid level (starting with the value at the height given by damp_level_1d, or possibly from the next grid point above), i.e.
km(k+1) = 1.1 * km(k). The values ofkmare limited to 10 m2 s-1 at maximum.The value of damp_level_1d must be within the range
[0, zu(nzt+1)].This parameter only comes into effect if the 1d-model is switched on for the initialization of the 3d-model via initializing_actions = 'set_1d-model_profiles'.
data_output_during_spinup#
- Fortran Type: L
-
Default: .FALSE.
Parameter for switching on data output in the spinup phase (see spinup_time).
If set to .T., 2D, 3D, timeseries and profile data, as well as surface data will be output at regular intervals as prescribed by the user. There will be no output of masked data during the spinup phase.
For radiation calculations during spinup, a starting time is calculated based on the setting of origin_date_time, so that the spinup starts at origin_date_time - spinup_time. The run after the spinup phase then starts at origin_date_time. In the output data, the timestamps during spinup will show negative values and the data after the spinup phase have timestamps >= 0.0 s.
dissipation_1d#
- Fortran Type: C*20
-
Default: detering
Calculation method for the energy dissipation term in the TKE equation of the 1d-model.
See also mixing_length_1d.
Currently 3 choices are available:
-
as_in_3d_model
This value requires to set mixing_length_1d = 'as_in_3d_model', too. The dissipation is calculated as in the 3d-model using \(diss = (0.19 + 0.74 * l / l_{grid}) * e^{1.5} / l\), with \(e\) being the subgrid TKE, \(l\) the mixing length, and \(l_{grid}\) defined by the grid spacing (i.e. it depends on the grid spacing). Be aware, that this setting will not give realistic vertical profiles of the mean wind. It mainly serves for testing purposes.
-
detering
The energy dissipation is calculated as \(diss = 0.064 * e^{1.5} / l\), with \(e\) being the subgrid TKE and \(l\) the mixing length.
-
prognostic
Energy dissipation is calculated via prognostic equation. To be used if the model is run in RANS mode using a TKE-\(\epsilon\) closure (see documentation).
-
dp_external#
- Fortran Type: L
-
Default: .FALSE.
Switch to employ external pressure gradient as the driving force for the flow.
The external pressure gradient is controlled by parameters dp_smooth, dp_level_b and dpdxy.
dp_external = .T. requires conserve_volume_flow = .F.. It is normally recommended to disable the Coriolis force by setting omega = 0.0, since under presence of Coriolis force the large scale pressure gradient is normally given implicitly via the geostrophic wind (see ug_surface and vg_surface.
dp_level_b#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: m
Lower limit of the vertical range for which the external pressure gradient is applied.
This parameter only applies if dp_external = .T.. It must hold the condition
zu(0) <=dp_level_b<= zu(nz). It can be used in combination with dp_smooth = .T. to generate a constant-flux boundary layer well below dp_level_b if dp_level_b >> 0.0. The flow below dp_level_b is then purely driven by the shear stress at the top of the layer, which means that within the layer and under stationary conditions the vertical turbulent momentum flux is constant with height.There is no upper limit of the vertical range. The external pressure gradient is always applied up to the top of the model domain.
dp_smooth#
- Fortran Type: L
-
Default: .FALSE.
Vertically smooth the external pressure gradient using a sinusoidal smoothing function.
This parameter only applies if dp_external = .T.. It is useful in combination with dp_level_b >> 0.0 to generate a constant-flux boundary layer well below dp_level_b.
dpdxy#
- Fortran Type: R (2)
- Default: 0.0
-
SI-Unit: Pa m-1
Values of the external pressure gradient applied along
x- andy-direction.This parameter only applies if dp_external = .T.. It sets the pressure gradient values along
x- andy-direction. Negative values mean an acceleration, positive values mean deceleration along the respective direction. For example, dpdxy = -0.0002, 0.0002, drives the flow in positivex-direction and negativey-direction.Note:
Suitable values of the external pressure gradient that produce wind profiles with desired magnitudes usually have to be determined by test runs.
dt#
- Fortran Type: R
- Default: Depends on timestep_scheme, internally calculated
-
SI-Unit: s
Length of time step for the 3d-model.
By default, the length of the time step is automatically determined after each time step based on stability criteria (e.g. Courant Friedrichs Levy (CFL) criterion, diffusion criterion, etc.). The respectiv value is then used for the next step. The actual value depends on these criteria and the parameter cfl_factor, which in turn depends on timestep_scheme.
If dt is assigned a value, then the time step is fixed to this value throughout the whole run (whether it fulfills the time step criteria or not). However, changes are allowed for restart runs (dt can also be used as a runtime parameter).
In case that the automatically calculated time step meets the condition: dt < 0.00001 * dt_max, the simulation will be aborted. Such situations usually arise in case of any numerical problem / instability which causes a non-realistic increase of the wind speed.
For flat terrain and horizontally homogeneous setups with cyclic boundary conditions the automatically calculated time step may be enlarged by using a coordinate transformation (see galilei_transformation), in order to save CPU time.
Warning:
The simulation will crash in case of a fixed dt, if the given value violates the time step criteria at any time throughout the run. Even worse, the simulation may continue to run up to the end, producingNaNdata, if you have not switched on the floating point error detection by setting a respective compiler option (e.g.-fpe0for the Intel compiler). You should first run the case without setting dt, check the minimum time step througout the simulation, and then choose a [dt](#initialization_parameters--dt smaller than that minimum.
dt_pr_1d#
- Fortran Type: R
- Default: undefined
-
SI-Unit: s
Temporal interval of vertical profile output of the 1d-model.
Data is written in ASCII format to file LIST_PROFIL_1D. This parameter is only in effect if the 1d-model has been switched on for the initialization of the 3d-model via initializing_actions = 'set_1d-model_profiles'.
dt_run_control_1d#
- Fortran Type: R
- Default: 60.0
-
SI-Unit: s
Temporal interval of runtime control output of the 1d-model.
Data is written in ASCII format to file RUN_CONTROL. This parameter is only in effect if the 1d-model has been switched on for the initialization of the 3d-model via initializing_actions = 'set_1d-model_profiles'.
dx#
- Fortran Type: R
- Default: 1.0
-
SI-Unit: m
Horizontal grid spacing along the x-direction.
Along the x-direction, only an equidistant grid is allowed. It is recommended to use an isotropic grid within the turbulent boundary layer, or for those parts of the model domain where turbulence is expected, i.e. dx = dy = dz.
dy#
- Fortran Type: R
- Default: 1.0
-
SI-Unit: m
Horizontal grid spacing along the y-direction.
Along y-direction, only an equidistant grid is allowed. It is recommended to use an isotropic grid within the turbulent boundary layer, or for those parts of the model domain where turbulence is expected, i.e. dx = dy = dz.
dz#
- Fortran Type: R (10)
- Default: undefined
-
SI-Unit: m
Vertical grid spacing.
At least one value must be assigned. It is recommended to use an isotropic grid within the turbulent boundary layer, or for those parts of the model domain where turbulence is expected, i.e. dx = dy = dz.
By default, the model uses constant grid spacing along the z-direction, but it can be stretched using either the deprecated parameters dz_stretch_level, dz_stretch_factor, dz_max, or the new ones dz_stretch_level_start, dz_stretch_level_end which allow grid stretching to a finer or coarser grid and the definition of several stretching regions.
If the new stretching method is employed, as many dz values (+1) need to be specified as values are given via dz_stretch_level_end, e.g. if 4 values have been given for dz_stretch_level_end, 5 values need to be given for dz. This way, the model knows which grid spacing shall be used at the beginning and at the end of a grid stretching region that has been defined via dz_stretch_level_start and dz_stretch_level_end.
Assuming a constant dz, the scalar levels (
zu) are calculated as:zu(0) = 0.0, zu(1) = dz(1) * 0.5, zu(k) = zu(k-1) + dz(1).The w-levels lie half between them:
zw(k) = ( zu(k) + zu(k+1) ) * 0.5,except for k=0, where
zw(0) = 0.0.
dz_max#
- Fortran Type: R
- Default: 999.0
-
SI-Unit: m
Allowed maximum vertical grid spacing.
If the vertical grid is stretched (see dz_stretch_factor and dz_stretch_level), dz_max can be used to limit the vertical grid spacing to the given maximum value. The default value is 999.0 in order to prevent unrealistic larger vertical grid spacings and model domains.
Note:
This parameter is deprecated. Please see parameters dz_stretch_level_start and dz_stretch_level_end for the new method to steer the grid stretching.
dz_stretch_factor#
- Fortran Type: R
-
Default: 1.08
Stretch factor for a vertically stretched grid (see dz_stretch_level).
The stretch factor should not exceed a value of approx. 1.10 - 1.12, otherwise the discretization errors due to the stretched grid are not negligible anymore (see Kalnay de Rivas, 1972).
Note:
This parameter is deprecated. Please see parameters dz_stretch_level_start and dz_stretch_level_end for the new method to steer the grid stretching.
dz_stretch_level#
- Fortran Type: R
- Default: undefined
-
SI-Unit: m
Height level above/below which the grid is to be stretched vertically.
dz_stretch_level is the height level above which the grid is to be stretched vertically. The vertical grid spacings dz above this level are calculated as
dz(k+1) = dz(k) * dz_stretch_factor
and used as spacings for the scalar levels (zu). The w-levels are then defined as:
zw(k) = ( zu(k) + zu(k+1) ) * 0.5.For ocean mode simulations, dz_stretch_level is the negative height level (in m) below which the grid is to be stretched vertically. The vertical grid spacings dz below this level are calculated correspondingly as
dz(k-1) = dz(k) * dz_stretch_factor.
Note:
This parameter is deprecated. Please see parameters dz_stretch_level_start and dz_stretch_level_end for the new method to steer the grid stretching.
dz_stretch_level_start#
- Fortran Type: R (9)
- Default: undefined
-
SI-Unit: m
Height level above/below which the grid is to be stretched vertically.
Up to 9 heights (separate stretching regions) are allowed. By default no stretching is applied.
dz_stretch_level_start is the height level above which the grid is to be stretched vertically. The vertical grid spacings dz between this level and the corresponding dz_stretch_level_end are calculated as
dz(k+1) = dz(k) * dz_stretch_factor_array
and used as spacings for the scalar levels
zu. The w-levels are then defined as:zw(k) = ( zu(k) + zu(k+1) ) * 0.5For ocean mode simulations, dz_stretch_level_start is the negative height level (in m) below which the grid is to be stretched vertically. The vertical grid spacings dz between this level and the corresponding dz_stretch_level_end are calculated as
dz(k-1) = dz(k) * dz_stretch_factor_array.
For each dz_stretch_level_start a corresponding dz_stretch_level_end and a dz must be defined, except for the last level. Here, it is possible to omit the value for dz_stretch_level_end to consider 'endless' stretching until the value of dz_max is reached. In that case the stretching factor can not be calculated and is set to the value of dz_stretch_factor.
Example:
dz = 10.0, 50.0, 100.0,
dz_stretch_level_start = 800.0, 1500.0,
dz_stretch_level_end = 1000.0, 3000.0,Here the vertical grid spacing starts with 10.0 m at the surface up to 800.0 m, then stretches to 50.0 m between 800.0 m and 1000.0 m. Up to a height of 1500.0 m it is constant again (50.0 m), and between 1500.0 m and 3000.0 m it is stretched to 100.0 m. Above 3000.0 m it has a constant value of 100.0 m.
Note that if the number of vertical grid points given via nz is not sufficiently large, the grid stretching mechanism may stop at a height smaller than the last value that is given by dz_stretch_level_end.
Hint:
dz_stretch_factor_arrayis an array defined in the code which stores the different stretching factors that correspond to the defined stretching regions. The factors are calculated internally except for the case when the old stretching with dz_stretch_factor, dz_stretch_level, and dz_max is used.
dz_stretch_level_end#
- Fortran Type: R (9)
- Default: undefined
-
SI-Unit: m
Height level until which the grid is to be stretched vertically.
Up to 9 heights (separate stretching regions) are allowed.
For each dz_stretch_level_end a corresponding dz_stretch_level_start and a dz must be defined. For further information see dz_stretch_level_start.
e_init#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: m2 s-2
Initial subgrid-scale TKE.
For e_init > 0.0, this parameter defines the initial subgrid-scale TKE level from which the initial eddy diffusivities Km and Kh are calculated. The parameter has an effect only if km_constant is not set. The setting only affects the spinup phase of the simulation.
Setting e_init may help to avoid very small timesteps that may appear right at the beginning of RANS-mode simulations with turbulence_closure = 'tke-e'.
e_min#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: m2 s-2
Minimum subgrid-scale TKE.
This option adds artificial viscosity to the flow by ensuring that the subgrid-scale TKE does not fall below a minimum threshold value as given by e_min. It may help to avoid that diffusivities become unphysically small in RANS-mode simulations with turbulence_closure = 'tke-e'. Similar internal thresholds are often used in other RANS models, too.
end_time_1d#
- Fortran Type: R
- Default: 864000.0
-
SI-Unit: s
Time to be simulated for the 1d-model.
The default value corresponds to a simulated time of 10 days. Usually, after such period inertial oscillations have completely decayed and the solution of the 1d-model becomes stationary (see damp_level_1d). This parameter only comes into effect if the 1d-model is switched on for initialization of the 3d-model via initializing_actions = 'set_1d-model_profiles'.
ensemble_member_nr#
- Fortran Type: I
-
Default: 0
Seed to vary the number sequence given by the chosen random-number generator.
By setting this parameter to an integer between 1 and 2000, PALM is able to generate statistically independent members based on the same simulation setup. For each ensemble member the simulated instantaneous turbulent flow fields completely differ, but their mean statistical properties would converge to the same value, if the simulations run long enough, and provided that a stationary state can be reached. For each different value of ensemble_member_nr a different set of random perturbations is imposed onto the flow fields.
This parameter becomes only effective, if random_generator = 'random-parallel' is set.
fft_method#
- Fortran Type: C*20
-
Default: temperton-algorithm
FFT-method to be used.
This option becomes only effective for psolver = 'poisfft' and defines which fast Fourier transformation (FFT) is used for solving the perturbation pressure equation. The respective FFT is also used for calculating power spectra (see spectra).
Two internal FFT methods are available (i.e. hard coded in the model), as well as FFT methods from external libraries.
Currently 4 choices are available:
-
temperton-algorithm
The Temperton-method from Clive Temperton (ECMWF) which is computationally efficient. In case of cyclic horizontal boundary conditions (see bc_lr, bc_ns) the number of horizontal grid points (nx+1, ny+1) to be used with this method must be composed of prime factors 2, 3 and 5. In case of non-cyclic horizontal boundary conditions nx,ny must be composed of prime factors 2, 3 and 5.
-
singleton-algorithm
The Singleton-method which is much slower than the Temperton-method, but has no restrictions concerning the number of grid points to be used with. Due to its very poor computational performance, this method is only recommended to be used for comparisons with the other available FFT methods.
-
fftw
The "Fastest Fourier Transform in the West". The FFTW-routines are significantly faster than those of the Temperton-FFT.
To use this method requires setting of the cpp-preprocessor switch
-D__fftwin the%cpp_optionsline of the configuration file, and to give the paths to the respective fftw include file and library via the%compiler_optionsand%linker_optionsline, respectively. In case the PALM installer has detected an existing FFTW library on your computer system, it automatically adds the required settings to the configuration file.Note:
The FFTW manual says:FFTW is best at handling sizes of the form \(2^a 3^b 5^c 7^d 11^e 13^f\) , where \(e + f\) is either 0 or 1, and the other exponents are arbitrary. Other sizes are computed by means of a slow, general-purpose algorithm (which nevertheless retains O(n log n) performance even for prime sizes.
In case of cyclic horizontal boundary conditions (see bc_lr, bc_ns) please check the size of nx+1 and ny+1, respectively. For non-cyclic horizontal boundary conditions nx,ny should match the above conditions.
-
system-specific
For running the code on NVidia GPUs, a CUDA-FFT is available, which requires setting of preprocessor switch
-D__cuda_fftin the%cpp_optionsline of the configuration file. Be aware that running the code on GPUs is a test feature, and that only the dynamic core of the model is able to run on this architecture. Contact the PALM developers for more information.
-
flux_input_mode#
- Fortran Type: C*40
-
Default: application-specific
Parameter to choose unit of fluxes that are given as input data.
Currently 3 choices are available:
-
application-specific
The flux input representation is chosen depending on the application. If no land surface model or urban surface model is used, and, at the same time, approximation = 'boussinesq', the fluxes are considered as "kinematic". For all other applications, the fluxes are represented as 'dynamic'.
-
dynamic
The flux input data is assumed to be given in "dynamic" units with W m-2 for sensible and latent heat fluxes, and N m-2 for momentum fluxes.
-
kinematic
The flux input data is assumed to be given in "kinematic" units, with K m s-1 for sensible heat fluxes, kg kg-1 m s-1 for latent heat fluxes, and m2 s-2 for momentum fluxes.
-
flux_output_mode#
- Fortran Type: C*40
-
Default: application-specific
Parameter to choose the unit of fluxes that appear in the output data.
Currently 3 choices are available:
-
application-specific
The flux output representation is chosen depending on the application. If no land surface model or urban surface model is used, and, at the same time, approximation = 'boussinesq', the fluxes are considered as "kinematic". For all other applications, the fluxes are represented as 'dynamic'.
-
dynamic
The flux output data is assumed to be given in "dynamic" units with W m-2 for sensible and latent heat fluxes, and N m-2 for momentum fluxes.
-
kinematic
The flux output data is assumed to be given in "kinematic" units with K m s-1 for sensible heat fluxes, kg kg-1 m s-1 for latent heat fluxes, and m2 s-2 for momentum fluxes.
-
galilei_transformation#
- Fortran Type: L
-
Default: .FALSE.
Switch for applying the Galilei-transformation to the coordinate system of the model.
galilei_transformation = .T. switches on a so-called Galilei-transformation which ensures that the coordinate system of the model moves with the speed and direction of the geostrophic wind. Alternatively, the horizontally averaged wind can be used as translation speed (see use_ug_for_galilei_tr. This speed will naturally change in time. The lower relative velocities in the translated system permit a larger time step dt. Moreover, with this method, numerical inaccuracies of the advection-scheme (concerns, in particular, the momentum advection) are minimized. Switching the transformation on is only worthwhile if the geostrophic wind (ug, vg) and the averaged horizontal wind clearly deviate from the value 0. In each case, the distance the coordinate system has been moved over time is written to file RUN_CONTROL.
Usage of the Galilei-transformation requires topography = 'flat', a homogeneous surface, and cyclic boundary conditions along
xandy.
homogenize_surface_temperature#
- Fortran Type: L
-
Default: .FALSE.
Parameter to switch on the homogenization of surface temperature at the beginning of a run.
At the beginning of a run, the surface temperature will be averaged over all bottom surface grid points in the simulated domain. This action will be carried out at the start of the main run as well as at the beginning of each restart run. The parameter is useful if the surface boundary condition for temperature changes between runs, more precisely, when the surface boundary condition changes from bc_pt_b = 'neumann', where the surface temperature might be spatially heterogeneous, to 'dirichlet'. This parameter is only allowed in combination with neutral = .F. and bc_pt_b = 'dirichlet'.
Note that in a nested run homogenize_surface_temperature must be set the same for all domains. For such runs the spatially mean surface temperature of the root domain defines the surface temperature of the child domains too. homogenize_surface_temperature will be automatically set to the root domain value.
homogenize_surface_temperature is allowed to be set as runtime parameter as well. In case of a restart run, it then overwrites the setting from the previous run and the new value will be used for further restarts, as long as no new setting via the runtime parameter namelist is made.
humidity#
- Fortran Type: L
-
Default: .FALSE.
Parameter to switch on the prognostic equation for the water vapor mixing ratio
q.The initial vertical profile of
qcan be set via parameters q_surface, q_vertical_gradient and q_vertical_gradient_level. Boundary conditions can be set via bc_q_b, bc_q_t, q_surface_initial_change and surface_waterflux.If the bulk cloud model is switched on (see bulk_cloud_model = .T.),
qbecomes the total water mixing ratio (sum of water vapor and liquid water). The bulk cloud model physics must be steered via bulk cloud parameters.
implicit_diffusion_1d#
- Fortran Type: L
-
Default: .FALSE.
Switch to calculate the diffusion terms in the prognostic equations of the 1d-model via the Crank-Nicolson time step scheme.
Using an implicit time step scheme as Crank-Nicolson allows for larger time steps than required by the diffusion time step criterion \(\Delta t \le \frac{\Delta z \Delta z}{K_m}\). The length of the time step can be controlled via implicit_timestep_factor.
implicit_timestep_factor#
- Fortran Type: R
-
Default: 5.0
Factor by which the time step is enlarged when using the Crank-Nicolson scheme.
The default value of the factor is a conservative one, that always should work. Testing larger values up to 40.0 is recommended, but results should be carefully compared with those that have been generated with the default value. Maximum possible values depend on the thermodynamic stability (temperature gradient) and the chosen turbulence parameterization.
inflow_disturbance_begin#
- Fortran Type: I
-
Default: Depends on bc_lr, bc_ns
Lower index limit of the horizontal range for which random perturbations are to be imposed on the horizontal velocity.
In case of cyclic horizontal boundary conditions, perturbations are added to the horizontal velocity components for each horizontal grid point. The value for inflow_disturbance_begin must not be smaller than zero or larger than nx (in case of non-cyclic lateral boundary conditions along the x-direction) or ny (in case of non-cyclic lateral boundary conditions along the y-direction).
If non-cyclic lateral boundary conditions are used, inflow_disturbance_begin defines the number of grid points (counted horizontally from the respective inflow plane), from which on perturbations are imposed to the horizontal velocity components, provided that perturbations have been switched on via parameter create_disturbances. The default value is then inflow_disturbance_begin =
MIN( 10, nx/2 or ny/2).In case of non-cyclic lateral boundary conditions the purpose of these disturbances is to continously trigger the onset of turbulence at the laminar inflow. Disturbances are not required in general, e.g., if turbulence recycling (see turbulent_inflow_method = 'recycle_...') is used.
inflow_disturbance_end#
- Fortran Type: I
-
Default: Depends on bc_lr, bc_ns
Upper index limit of the horizontal range for which random perturbations are to be imposed on the horizontal velocity.
In case of cyclic horizontal boundary conditions, perturbations are added to the horizontal velocity components for each horizontal grid point. The value for inflow_disturbance_end must not be smaller than zero or larger than nx (in case of non-cyclic lateral boundary conditions along the x-direction) or ny (in case of non-cyclic lateral boundary conditions along the y-direction).
If non-cyclic lateral boundary conditions are used, inflow_disturbance_end defines the number of grid points (counted horizontally from the respective inflow plane) up to which perturbations are imposed to the horizontal velocity components, provided that perturbations have been switched on via parameter create_disturbances. The default value is then inflow_disturbance_end =
MIN( 100, 3/4 nx or 3/4 ny).In case of non-cyclic lateral boundary conditions the purpose of these disturbances is to continously trigger the onset of turbulence at the laminar inflow. Disturbances are not required in general, e.g., if turbulence recycling (see turbulent_inflow_method = 'recycle_...') is used.
initializing_actions#
- Fortran Type: C*100
-
Default: undefined
Initial conditions to be set and respectivly required actions to be carried out at model start.
This parameter does not have a default value and must be assigned in each parameter set. For restart runs, initializing_actions = 'read_restart_data' must be set. The available settings for the initial run (of a possible job chain) are described in the following.
Values may be combined, separated by one or more blanks, e.g. initializing_actions = 'set_constant_profiles initialize_vortex' , but the values of 'set_constant_profiles' , 'set_1d-model_profiles' , 'cyclic_fill' , 'read_restart_data' , 'read_from_file' and 'by_user' must not be given at the same time.
For child domains in inital runs initializing_actions = 'interpolate_from_parent' is automatically set.
Currently 10 choices are available:
-
by_user
The initialization of the arrays of the 3d-model and surface-related quantities is under complete control of the user and has to be done in routine
user_init_3d_modelof the user-interface. Only recommended for very experienced users. -
cyclic_fill
3d-data from a precursor run are read. The precursor run is allowed to have a smaller domain along
xandycompared with the main run. Also, different numbers of processors can be used for these two runs. Limitations are that the precursor run must use cyclic horizontal boundary conditions and that the number of vertical grid points, nz, must be the same for the precursor run and the main run. If the total domain of the main run is larger than that of the precursor run, the domain is filled by cyclic repetition of the (cyclic) precursor data. This initialization method is required if turbulence recycling is used (see turbulent_inflow_method = 'recycle_...'). 3d-data must be made available to the run by adding the activation string 'cyclic' topalmrun-option-a, e.g.-a 'd3# cyclic'. The usage of a turbulent inflow is explained here. Note that in case of reference_state = 'initial_profile', the main run uses the respective initial profiles of the precursor run. -
initialize_bubble
A 2d-Gauss-like shaped perturbation (y,z) of 0.4 K is added to the initial temperature field at a height of 150 m and centered along the y-direction. The temperature perturbation decreases with a standard deviation of 300 m and 150 m along
yandz, respectively. This is a simple method to induce a rising warm air bubble producing clouds in a humid environment. -
initialize_ptanom
An option for testing scalar advection schemes.
A 2d-Gauss-like shape disturbance (x,y) is added to the initial temperature field with radius 10.0 * dx and centered at
ic =(nx+1)/2. Scalar advection tests may also require a horizontal wind profile constant with height and diffusion switched off (see 'initialize_vortex' ). Additionally, the buoyancy term must be switched off in the equation of motion for w (this requires the user to comment out the call of buoyancy in the source code ofprognostic_equations.f90). -
initialize_vortex
An option for testing advection schemes.
The initial velocity field of the 3d-model corresponds to a Rankine-vortex with a vertical axis. Free-slip boundary conditions for u and v (see bc_uv_b, bc_uv_t) are required. In order not to distort the vortex, an initial horizontal wind profile constant with height is necessary (to be set via initializing_actions = 'set_constant_profiles') and some other conditions have to be met (neutral stratification, diffusion must be switched off, see km_constant). The center of the vortex is located at
ic =(nx+1)/2. It extends from k = 0 to k = nz+1. Its radius is 8 * dx and the exponentially decaying part extends to 32 * dx (see source code routineinit_rankine.f90). -
interpolate_from_parent
This option is used for nested domains in nested initial runs. The initial conditions for a nested domain are then interpolated from its parent domain. This value is automatically set for child domains in initial runs.
-
read_from_file
Initialization with input data derived from external models. For the DWD-COSMO model, an escpecially designed preprocessor tool called INIFOR is available. This method requires a dynamic input file. This option can be combined with other options.
-
set_constant_profiles
A horizontal velocity profile consisting of linear sections (see ug_surface, ug_vertical_gradient, ug_vertical_gradient_level and vg_surface, vg_vertical_gradient, vg_vertical_gradient_level, respectively) as well as a vertical temperature and humidity profile consisting of linear sections (see pt_surface, pt_vertical_gradient, q_surface and q_vertical_gradient) are assumed as horizontally homogeneous initial state. The subgrid-scale TKE is set to 0 but eddy diffusivities
kmandkhare set to very small values because otherwise no TKE would initially be generated.Instead of using the geostrophic wind for constructing the initial u,v-profiles, these profiles can also be directly set using parameters u_profile, v_profile, and uv_heights, e.g. if observed profiles are available as initial state. In runs with non-cyclic horizontal boundary conditions these profiles are also used as fixed mean inflow profiles.
-
read_restart_data
Must be set in restart runs, for parent as well as for all existing child domains.
-
read_spinup_data
Initialization of land and wall surface data with provided spinup data from a previous run. This option is thought to initialize surfaces in an initial run without repeating the surface spinup. This is e.g. useful in ensemble runs (see ensemble_member_nr) where the initial state of the soil, the walls, the surfaces and the atmosphere is identical but the realization of random turbulence is different.
Values may be combined with all other actions, e.g. initializing_actions = 'set_constant_profiles', except for the values of 'read_restart_data' . To use this option, spinup data from a previous run with spinup_time /= 0.0 must be available. This data is written into SPINOUT triggered by
palmrun ... -a "... spinout". Please note, reading and writing surface spinup data is only possible with restart_data_format = 'mpi' or 'mpi_shared_memory'. In order to input the surface data in an initial run, the spinin attribute must be given:palmrun ... -a "d3# ... spinin".
-
km_constant#
- Fortran Type: R
- Default: undefined
-
SI-Unit: m2 s-1
Constant eddy diffusivities are used.
By specifying this parameter, both the 1d and the 3d-model use constant values (in space and in time) for the eddy diffusivities
km =km_constant andkh = km /prandtl_number. The prognostic equation for the subgrid-scale TKE is switched off. Constant eddy diffusivities are only allowed with the constant flux layer (constant_flux_layer) switched off. In DNS mode km_constant can be used to set the molecular viscosity.
large_scale_forcing#
- Fortran Type: L
-
Default: .FALSE.
Parameter to choose large-scale forcing from an external file.
large_scale_forcing = .T. requires the input file LSF_DATA. This file contains two kinds of information: time-dependent surface values and time-dependent profiles which can be provided either by measurements or by larger scale models. In more detail, the file contains the following data/parameters:
- time-dependent surface heat flux
- time-dependent surface water flux
- surface temperature (see pt_surface
- surface humidity
- surface pressure (see surface_pressure)
- vertical profiles of the geostrophic wind components (see ug and vg)
- the large-scale vertical subsidence profile
w_subs - the horizontal large-scale advection tendencies of temperature
td_lsa_thetaland humiditytd_lsa_q - the large-scale subsidence tendencies of temperature
td_sub_thetaland humiditytd_sub_q
An example can be found here.
large_scale_forcing = .T. requires humidity = .T.. It is not implemented for the ocean mode, and it does also not work for non-cyclic lateral boundary conditions and non-flat topography. It is possible to drive the simulations either by means of surface fluxes or by means of prescribed surface values for temperature and humidity.
In case of simulating moderately tall buildings on otherwise flat terrain, large-scale forcing can be used with additional setting of lsf_exception = .T.. However, such setups havn't been tested so far and results should be checked with care.
In case that large-scale forcing shall be used without nudging, initial profiles of potential temperature, humidity, and horizontal wind components have to be provided by means of pt_surface, pt_vertical_gradient, pt_vertical_gradient_level etc.
large_scale_subsidence#
- Fortran Type: L
-
Default: .FALSE.
Parameter to enable large-scale subsidence/ascent.
Calculates and adds an additional tendency term
tend_substo the scalar quantities prognostic equations. For potential temperature, it e.g. looks liketend_subs(k,j,i) = - w_subs(k)* dpt(k,j,i) / dz.The profile for the subsidence velocity
w_subscan either be set via subs_vertical_gradient and subs_vertical_gradient_level or by reading it from the large-scale forcing data set LSF_DATA.In case of use_subsidence_tendencies = .T., the subsidence velocity
w_subsis not used. Instead, subsidence tendencies for temperature and humidity are taken from the large-scale forcing data set LSF_DATA and applied to the respective prognostic variables.large_scale_subsidence is not implemented for the ocean mode.
Attention:
The large-scale vertical motion is only applied to the prognostic equation for the scalar quantities (potential temperature, humidity if humidity = .T., or passive scalar if passive_scalar = .T.). It must not be applied to the momentum equations because it would violate the incompressibility condition. Applying it to the horizontal velocity components would result in mass inconsistencies.
latitude#
- Fortran Type: R
- Default: 55.0
-
SI-Unit: degree
Geographical latitude.
The value of this parameter determines the value of the Coriolis parameters
fandf*, provided that the angular velocity (see omega) is non-zero. Note that at equator the horizontal component of the Coriolis force is zero, while its vertical component has its maximum, and vice versa at the poles. For completely switching off the Coriolis force, it is thus recommended to set omega = 0.0 instead.latitude is also used by the radiation model to determine the position of the sun.
If a static driver is used, latitude will be overwritten by the value given in the static driver.
longitude#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: degree
Geographical longitude.
The value is used by the radiation model to determine the position of the sun.
If a [static driver](https://palm.muk.uni-hannover.de/trac/wiki/doc/app/iofiles/pids/static is used, longitude will be overwritten by the value given in the static driver.
loop_optimization#
- Fortran Type: C*16
-
Default: cache
Method used to optimize loops within the prognostic equations.
Currently 2 choices are available:
-
cache
All prognostic equations are solved within one big loop over the two horizontal indices
iandjfor a good cache utilization. This setting is recommended for all Intel/AMD/ARM based processors. -
vector
Single 3d-loops are used to calculate each tendency term of each prognostic equation. This allows for long vectors and is recommended for vector processors (e.g. NEC, but also for GPUs).
-
lsf_exception#
- Fortran Type: L
-
Default: .FALSE.
Parameter to explicitly allow large-scale forcing in case of topography /= 'flat'.
So far, large_scale_forcing is not implemented to be used together with topography. In case of simulating moderately tall buildings on otherwise flat terrain, large-scale forcing can be used with additional setting of lsf_exception = .T.. However, such setups havn't been tested so far and results should be checked with care.
masking_method#
- Fortran Type: L
-
Default: .FALSE.
Switch for topography pressure boundary conditions in case of psolver = 'multigrid_noopt'.
In case of topography, by default, the pressure solvers (see psolver) are running 'through' the topography, which means that no pressure boundary conditions are considered at the respective topography/wall surfaces (topography is masked).
For psolver = 'multigrid_noopt' Neumann boundary conditions for perturbation pressure are used by default at all wall boundaries. In case of masking_method = .T., this solver runs through the topography like it is done for all other pressure solvers that are available.
Note:
masking_method only has an effect if [psolver] (#initialization_parameters--psolver) = 'multigrid_noopt', otherwise masking is always used.Remarks:
In case of very complex topography, including narrow street canyons or complex shapes resolved by only a few grid points, the use of psolver = 'multigrid_noopt' in combination with masking_method = .F. can lead to a blow up of wind velocity. A possible workaround to prevent such velocity blow-up is to preprocess the topography by filling holes and removing complex shapes on the small scale before running the LES. (This approach should have no significant effect on the flow field, as in LES the flow in such regions is poorly resolved and do not yield to reliable physical information.)If topography is prescribed and psolver = 'multigrid_noopt', the mean velocity-divergence reduction (see RUN_CONTROL) is smaller in case of masking_method = .T. compared to .F. (about one-order of magnitude). This smaller reduction can be attributed to wall-bounded grid points, where the divergence after pressure correction remains significantly larger in case of masking, while the divergence reduction for non-wall-bounded grid points is similar in both cases. However, validation tests revealed that this smaller divergence reduction has no significant effect on the resulting flow.
mg_cycles#
- Fortran Type: I
-
Default: 4
Number of cycles to be used with the multigrid scheme.
This parameter determines the number of cycles to be carried out in the multigrid method used for solving the Poisson equation for perturbation pressure (see psolver). The type of the cycles can be set with cycle_mg.
By default, a fixed number of cycles (4) is carried out. This number of cycles might not be sufficient for every simulation setup! The user should carefully check the RUN_CONTROL file whether the divergence of the velocity field is sufficiently reduced by the pressure solver. It should be reduced at least by two orders of magnitude during each time step. For cyclic boundary conditions along both horizontal directions (see bc_lr and bc_ns) mg_cycles = 2 is typically a good choice, for non-cyclic lateral boundary conditions or runs with topofraphy mg_cycles = 4 may be sufficient.
If set mg_cycles = -1, the number of cycles depends on the requested accuracy of the scheme (see residual_limit) and may vary from time step to time step. In this case, the CPU time for a run will be difficult to estimate since it heavily depends on the total number of the cycles to be carried out.
mg_switch_to_pe0_level#
- Fortran Type: I
-
Default: -1
Multigrid level at which data shall be gathered on PE0.
In case of a parallel run and psolver = 'multigrid' or 'multigrid_noopt', the value of this parameter defines on which multigrid level the data are gathered on PE0 in order to allow for a further coarsening of the grid. The finest grid defines the largest grid level. If mg_switch_to_pe0_level = 0, the gathering level is determined automatically and displayed in file RUN_CONTROL. It is only possible to gather data from a level larger than the one determined automatically. A test run may be necessary to determine this level.
Default setting is mg_switch_to_pe0_level = -1, which prevents that data are collected on PE0 at all, i.e. coarsening of grids is limited by what the subdomains allow.
Note:
The default setting is significantly faster compared to automatically determining or explicitly prescribing mg_switch_to_pe0_level. Even though the divergence reduction of the multigrid solver is slightly smaller with the default setting, gathering on PE0 does not justify the additional cost so far.
mixing_length_1d#
- Fortran Type: C*20
-
Default: blackadar
Mixing length used in the 1d-model.
See also dissipation_1d.
Currently 2 choices are available:
-
blackadar
The so-called Blackadar mixing length is used, where \(l = \frac{\kappa z}{1 + \frac{\kappa z}{\lambda}}\), with the limiting value of \(\lambda = 2.7E^{-4} \frac{u_g}{f}\). This is a typical approach used in ensemble-averaged (RANS) models, which provides realistic vertical profiles of the mean wind for a neutrally stratified boundary layer. Such profiles can be used as first guess for the initial condition of the 3d-model.
-
as_in_3d_model
The mixing length is calculated as in the 3d-model (i.e. it depends on the grid spacing). Be aware, that this setting will not give realistic vertical profiles of the mean wind. It mainly serves for testing purposes.
-
momentum_advec#
- Fortran Type: C*16
-
Default: ws-scheme
Advection scheme to be used for the momentum equations.
Currently 3 choices are available:
-
ws-scheme
The 5th order upwind scheme of Wicker and Skamarock (2002, Mon. Wea. Rev, 130, 2088-2097) is used. It implies a small numerical dissipation that stabilizes the solution. Due to the additional dispersion, small scale fluctuations \(< 8 \Delta\) are strongly damped, visible as a strong drop-off in the energy spectrum for the largest wavenumbers. The dispersion error is small. To assure a stable numerical solution the time integration has to be carried out with timestep_scheme = 'runge-kutta-3'.
The scheme is based on a formulation of the advection term in flux form, which requires the flow field to be free of divergence, else a stable numerical solution is not given. Take care, that the pressure solver sufficiently reduces the divergence, especially if psolver = 'multigrid' has been set. call_psolver_at_all_substeps = .F. should not be set.
The computation of resolved-scale turbulent fluxes for output purposes is done inside the advection scheme, in order to provide data that is consistent with the numerical flow solution. Calculating turbulent fluxes via turbulent fluctuations that are defined as the deviation of a quantity from the horizontal domain average (like usually done after a time step within the PALM routine
flow_statistics) may give completely wrong results.Note: Due to the larger stencil of this scheme (which extends over seven grid points) vertical grid stretching should be limited. With respect to the stencil and the domain decomposition, the scheme requires three ghost layers at the subdomain boundaries along x and y, so small subdomains (less than about \(15 \times 15\) grid points along x and y) will require an excessively large number of ghost points. As a consequence, the time for exchanging ghost point data via the communication network may drastically increase and so decrease the computational performance.
-
pw-scheme
The 2nd order scheme of Piacsek and Williams (1970, J. Comp. Phys., 6, 392-405) with central differences in the form C3 is used. The drop-off in the energy spectrum is not as strong as for momentun_advec = 'ws-scheme', but the dispersion error is much stronger. In the vicinity of strong gradients, the scheme of Piacsek and Williams tends to produce so-called \(2\Delta\) wiggles, which are well visible in results, e.g. at building corners.
-
up-scheme
1st order upwind scheme. The upstream scheme guarantees a stable solution only in combination with timestep_scheme = 'euler'. Note, this scheme is very diffusive, and hence, not well-suited for turbulence-resolving simulations.
-
monotonic_limiter_z#
- Fortran Type: L
-
Default: .FALSE.
Control flag that enables a monotonic flux limiter for the vertical advection of passive scalars according to Skamarock (2006, Mon. Wea. Rev 134, 2241-2250).
If the limiter is enabled, vertical scalar advection will be monotone and positive definite but strong gradients may be smoothed. This will avoid numerical oscillations (over-/undershootinng) that can e.g. cause unrealistically high concentrations within poorly resolved cavities within urban environments, e.g. narrow street canyons, which can spoil the entire simulation results.
Please note, that the limiter is only effective up to the height of the highest topography, and if scalar_advec = 'ws-scheme' is set. Moreover, the monontonic limiter is currently only implemented for loop_optimization = 'cache'.
netcdf_precision#
- Fortran Type: C*20 (11)
-
Default:
_NF90_REAL4 Defines the accuracy of the netCDF output.
By default, all netCDF output data has single precision (4 byte) accuracy. Double precision (8 byte) can be choosen alternatively. Accuracy for the different output data (cross sections, 3d-volume data, spectra, etc.) can be set independently by substituting the placeholder
<out>with a string from the following list:string meaning 'xy' horizontal cross section 'xz' vertical (xz) cross section 'yz' vertical (yz) cross section '2d' all cross sections '3d' volume data 'pr' vertical profiles 'ts' time series, particle time series 'sp' spectra 'prt' particles 'all' all output quantities Example:
If all cross section data and the particle data shall be output in double precision and all other quantities in single precision, set netcdf_precision = '2d_NF90_REAL8', 'prt_NF90_REAL8'.Currently 2 choices are available:
-
_NF90_REAL4 single precision (4 byte)
-
_NF90_REAL8 single precision (8 byte)
-
neutral#
- Fortran Type: L
-
Default: .FALSE.
Parameter to switch off calculation of the prognostic equation for temperature.
For simulating flows with pure neutral stratification, solving the temperature equation can be switched off via neutral = .T. in order to save CPU time. Additionally, this will also switch off the calculation of all buoyancy related terms. In such case, non-zero values for parameters surface_heatflux and top_heatflux are not allowed.
ngsrb#
- Fortran Type: I
-
Default: 2
Number of Gauss-Seidel iterations to be carried out on each grid level of the multigrid Poisson solver.
In case of using the multigrid method for solving the Poisson equation for perturbation pressure (see psolver), this parameter defines the number of Gauss-Seidel iterations to be carried out on each grid level. High values of ngsrb give better convergence. The default value of 2 reduces the divergence of the preliminary velocity field by about 1-2 orders of magnitude, which is sufficient in most cases.
Note: The value of ngsrb has a significant effect on the CPU requirement of the run.
nsor#
- Fortran Type: I
-
Default: 20
Number of iterations to be used with the SOR-scheme.
This parameter is only effective if the SOR-scheme is selected as pressure solver (psolver = 'sor'). The number of iterations necessary for a sufficient convergence of the scheme depends on the total number of grid points and is to be determined by appropriate test runs (the default value will not at all be sufficient for larger grid point numbers). The number of iterations used for the first call of the SOR-scheme (at
t= 0) is determined via parameter nsor_ini.
nsor_ini#
- Fortran Type: I
-
Default: 100
Initial number of iterations with the SOR-scheme.
This parameter is only effective if the SOR-scheme is selected as pressure solver (psolver = 'sor') and specifies the number of initial iterations of the SOR scheme (at
t= 0). The number of subsequent iterations at the following time steps is determined via parameter nsor. Usually, nsor < nsor_ini, since subsequent calls of the iterative pressure solver use the solution of the previous call as the initial value. Suitable test runs should determine whether sufficient convergence of the solution is obtained with the default value, and if necessary the value of nsor_ini should be adjusted.
nudging#
- Fortran Type: L
-
Default: .FALSE.
Parameter to activate nudging towards a horizontal mean state.
Nudging is a relaxation technique which adjusts the large-eddy simulation to a given, larger scale flow situation. It can, for example, be used to simulate an observed situation. Further information can be found here.
With nudging = .T., additional tendencies are calculated for the prognostic variables
u,v,lpt, andq. It requires setting of humidity = .T. as well as large_scale_forcing = .T.. So far, it is not implemented for the ocean mode and non-cyclic lateral boundary conditions. An example can be found here.Additionally, for nudging = .T. the input file NUDGING_DATA has to be provided. This file contains profile information at several time steps about the relaxation time scale \(\tau\) and the prognostic variables
u,v,w,lpt,qwhich must be provided by a larger scale model or by measurements.
nx#
- Fortran Type: I
-
Default: undefined
Number of gridpoints in x-direction.
A value for this parameter must be assigned. Since the lower array bound in PALM starts with i = 0, the actual number of grid points is equal to nx+1. In case of cyclic boundary conditions along x, the domain size is (nx+1)* dx.
nx+1 must be even in case of cyclic boundary conditions along x (see bc_lr) if the fft-solver is used for calculating the Poisson equation (see psolver). The multigrid Poisson solver also requires nx+1 to be even if more than one PE is used for the run.
For coupled atmosphere-ocean runs (see details), the product of dx and nx in both parameter files PARIN and PARIN_O has to be same (same model domain length in x-direction).
ny#
- Fortran Type: I
-
Default: undefined
Number of gridpoints in y-direction.
A value for this parameter must be assigned. Since the lower array bound in PALM starts with j = 0, the actual number of grid points is equal to ny+1. In case of cyclic boundary conditions along y, the domain size is (ny+1) * dy.
ny+1 must be even in case of cyclic boundary conditions along y (see bc_nc) if the fft-solver is used for calculating the Poisson equation (see psolver). The multigrid Poisson solver also requires ny+1 to be even if more than one PE is used for the run.
For coupled runs (see details) the product of dy and ny in both parameter files PARIN and PARIN_O has to be same (same model domain length in y-direction).
nz#
- Fortran Type: I
-
Default: undefined
Number of gridpoints in z-direction.
A value for this parameter must be assigned. Since the lower array bound in PALM starts with k = 0 and since one additional grid point is added at the top boundary (k = nz+1), the actual number of grid points is nz+2. However, the prognostic equations are only solved up to nz (u, v, scalar quantities) or up to nz-1 (w). The top boundary for u, v and scalar quantities is at k = nz+1 (u, v) while at k = nz for w.
The multigrid Poisson solver requires nz to be even.
omega#
- Fortran Type: R
- Default: 7.29212e-05
-
SI-Unit: rad s-1
Angular velocity of the rotating system.
The angular velocity of the earth is set by default. The values of the Coriolis parameters are calculated as:
f = 2.0 * omega * sin(latitude)
f* = 2.0 * omega * cos(latitude)
omega_sor#
- Fortran Type: R
-
Default: 1.8
Convergence factor to be used with the SOR-scheme.
If the SOR-scheme is selected (psolver = 'sor'), this parameter determines the value of the convergence factor, where 1.0' <= omega_sor < 2.0. The optimum value of omega_sor depends on the number of grid points along the different directions in space. For non-equidistant grids, it can only be determined by appropriate test runs.
origin_date_time#
- Fortran Type: C*23
-
Default: 2019-06-21 12:00:00 +00
Date and time at model start.
Time is given as local time, and the last three characters define the distance (in hours) between local time and UTC, e.g. '14:30:00 +02' means that the local time is 2 hours ahead of UTC (which is then '12:30:00'). The given local time is internally converted to UTC. In output files, time is always given in UTC. The correct local solar position is calculated from UTC, latitude and longitude.
Time may also be given in UTC directly.
Examples:
Setting model-start local time to 14th May 1995 at 6:30 PST (American Pacific Standard Time):
origin_date_time = '1995-05-14 06:30:00 -08'
Same time, but using UTC:
origin_date_time = '1995-05-14 14:30:00 +00'See use_fixed_date and use_fixed_time for further time-related options.
Please see also possible side effects concerning the time-coordinates in the dynamic driver input file.
outflow_damping_factor#
- Fortran Type: R
-
Default: 0.0
Factor for damping the 3d-field of u, v, w and pt near the outflow boundary.
If a non-zero value is assigned to outflow_damping_factor, a Rayleigh-damping is applied to all velocity components and the potential temperature near the outflow boundary in case that non-cyclic lateral boundary conditions (bc_lr = 'dirichlet/radiation', bc_lr = 'radiation/dirichlet', bc_ns = 'dirichlet/radiation', or bc_ns = 'radiation/dirichlet') are used.
The method removes possible spurious jets that are directed into the domain, which may be produced by the radiation boundary condition if a wall normal velocity component at the outflow plane is directed into the domain. This may happen due to turbulent fluctuations or due to recirculations behind buildings/topography near the outflow. With this method the turbulent fluctuations or local recirculations will vanish so that no negative wall normal velocities can occur.
If switched on, u, v, w and pt are forced towards the respective value of its basic state (defined by the initial profile), similar to the Rayleigh-damping that can be applied at the domain top (see rayleigh_damping_factor) or the pt-damping that can be apllied at the inflow boundary (see pt_damping_factor). The intensity of the damping is controlled by the value of outflow_damping_factor. The damping starts weakly at a distance from the outflow boundary defined by outflow_damping_width and rises according to a sin2-function to its maximum value at the outflow.
If the outflow damping is used in combination with topography, then the terrain height must be zero at the inflow and outflow boundary in order to guarantee that inflow and outflow profiles are identical and the volume flow is conserved.
outflow_damping_width#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: m
Width of the outflow damping layer.
In case of non-cyclic lateral boundary conditions, this parameter determines the width of the damping region in which the outflow damping is applied. The damping region starts at a distance of outflow_damping_width upstream of the outflow boundary and ranges up to the outflow boundary. The intensity of the damping is given by outflow_damping_factor.
outflow_source_plane#
- Fortran Type: R
- Default: undefined
-
SI-Unit: m
Horizintal position of the vertical source plane from where instantaneous values of prognostic variables are copied to the outflow boundary.
Parameter is effective only in case of turbulent_outflow = .T.. Affected quantities are
u,v,w,pt,q,s, ande. The position needs to be given as the distance from the inflow boundary. The source plane must be positioned inside the model domain, i.e.dx<= outflow_source_plane <=nx*dx.
passive_scalar#
- Fortran Type: L
-
Default: .FALSE.
Parameter to switch on the prognostic equation for a passive scalar
s.The initial vertical profile of
scan be set via parameters s_surface, s_vertical_gradient, and s_vertical_gradient_level. Boundary conditions can be set via s_surface_initial_change and surface_scalarflux.
prandtl_number#
- Fortran Type: R
-
Default: 1.0
Ratio of the eddy diffusivities for momentum and heat (Km/Khf).
For runs with constant eddy diffusivity (see km_constant), this parameter can be used to assign the Prandtl number.
psolver#
- Fortran Type: C*16
-
Default: poisfft
Scheme to be used to solve the Poisson equation for the perturbation pressure.
Solution of the Poisson equation is required after each time step (or partial time steps in the Runge-Kutta schemes) in order to keep the velocity field free of divergence.
In principle, the so-called masking method is used, meaning that the schemes run 'through' the topography/buildings, so topography flags are completely ignored (i.e. not used) and pressure boundary conditions at walls are ignored (except at the bottom surface at grid point
k= 0). Divergence for grid points within topography/buildings are set to zero and wall normal velocities at surface grid points are set to zero after the pressure correction has been carried out. Because of that, the divergence reduction in the vicinity of walls is less effective. For psolver = 'multigrid_noopt' the masking method can be switched off with setting masking_method = .F.. However, in that case pressure boundary conditions at walls are currently not well defined, so using this setting is not recommended.In case of using a multistep Runge-Kutta time step scheme (see timestep_scheme), the Poisson equation is by default solved after each of the substeps. In order to speed-up performance, it may be solved only for the last substep (see call_psolver_at_all_substeps), but in general, the divergence reduction then might not be sufficient enough, resulting in a blow-up of velocities.
Currently 5 choices are available:
-
poisfft
Direct method using FFT along x and y, solution of a tridiagonal matrix along z, and backward FFT. The FFT routines to be used can be determined via the initialization parameter fft_method. The FFT solver can be used for cyclic boundary conditions both along x and y, and for non-cyclic boundary conditions both along x and y (e.g. in case of offline nesting using a dynamic driver). Mixed boundary conditions, e.g. cyclic along x and non-cyclic along y are not allowed. Depending on the FFT method and the horizontal boundary conditions that are used, the number of grid points along x and y must follow specific restrictions. Error messages are issued if restrictions are not met.
-
poisfft_sm
Same as psolver = 'poisfft', but the solver uses an internal 1d-decomposition for pressure. This way, the number of transpositions that are required between the forward/backward FFTs and the tridiagonal solver is reduced from 6 to 2, which for larger setups may improve the overall model performance by more than 20-30%. The virtual PE grid (npex, npey) for the 2d-decomposition (which is used for all other quantities) is calculated by the solver. The solver usually sets npey to the number of cores available on a node, and npex to the number of nodes that are used in a run. If this leads to an unfavorable ratio of npex / npey, cores on the node are split into smaller shared memory groups. Pressure is defined as a shared memory array, to which all cores on a node (or in a smaller shared memory group) have access. This way, only one transposition from x to y (and the inverse one) is required to solve the Poisson equation.
Attention: psolver = 'poisfft_sm' is not allowed if less than 5 cores are used in a run.
Runtime-parameters npex and npey are not allowed to be set.
The total number of cores (given via
palmrun-option-Xmust be a multiple of the number of cores used per node (given viapalmrun-option-T, and-Tmust be a multiple of 4. Be aware that if-Xis not a multiple if-T, the setting of-Tis usually ignored by the batch system, and replaced by a value of which a multiple just approximates the total number of cores, which is then usually not a multiple of 4.This solver may not work for extreme ratios of nx / ny.
Respective error messages will be issued in case that the above restrictions are violated.
-
multigrid
Iterative multigrid scheme. v- and w-cycles (see cycle_mg) are implemented. The convergence of the iterative scheme can be steered by the number of v-/w-cycles to be carried out for each call of the scheme (mg_cycles) and by the number of Gauss-Seidel iterations (see ngsrb) to be carried out on each grid level. Instead, the requested accuracy can be given via residual_limit. The smaller this limit is, the more cycles have to be carried out and the number of cycles may vary from time step to time step. Because of possible excessive CPU time demands using residual_limit is not recommended in general.
If mg_cycles is set to its optimal value, the computing time of the multigrid scheme amounts approximately to that of the direct solver 'poisfft' as long as the number of grid points in the three spatial directions corresponds to a power-of-two (2n) where n >= 5 must hold. With large n, the multigrid scheme can even be faster than the direct solver (although its accuracy is several orders of magnitude worse, but this does not affect the accuracy of the simulation). Nevertheless, test runs may have to be carried out in order to find out the optimum value for mg_cycles, because the CPU time of a run very critically depends on this parameter.
This scheme requires that the number of grid points of the subdomains (or of the total domain, if only one PE is used) along each of the directions can, at least, be divided once by 2 without rest. A further side condition arising from performance optimizations is that the number of grid points along z on the coarsest grid level must not be an odd number. If this is the case, the number of possible grid levels along z is reduced by one. For example, if nz(level 1) = 150 and nz(level 2) = 75 (odd number) it results in no possible grid coarsening (only one grid level possible), while nz(level 1) = 152, nz(level 2) = 76, nz(level 3) = 38, nz(level 4) = 19 results in 3 possible multigrid levels.
With parallel runs, starting from a certain grid level the data of all subdomains can be gathered on PE0 in order to allow for a further coarsening of the grid. The grid level for gathering can be manually set by mg_switch_to_pe0_level.
'multigrid' is highly optimized for better performance. Data along the
kindex (vertical direction) is sorted from alternate even and odd indices into two blocks of completely even and odd indices respectively, which allows vectorization on INTEL processors of loops in the red/black scheme. Additionally, topography flags are completely ignored (i.e. not used) meaning that setting of masking_method = .F. is ignored. On fine grid levels only even or odd data are send via the lateral ghost point exchange. Speedup of the multigrid solver for medium sized cases (1024*1024*192 grid points) can be about 40% compared to 'multigrid_noopt'. -
multigrid_noopt
Similar as 'multigrid', but less optimized. However, in contrast to 'multigrid', 'multigrid_noopt' allows masking_method = .F., i.e. setting of Neumann boundary conditions for the perturbation pressure at all wall boundaries.
Please note: In combination with masking_method = .F., the 'multigrid_noopt' can lead to a blow-up of wind velocity in case of narrow street canyons or complex building shapes resolved by only a few grid points, because of over-determination of perturbation pressure. Please see further remarks.
-
sor
Successive over-relaxation method (SOR). The convergence of this iterative scheme is steered with the parameters omega_sor, nsor_ini and nsor.
Compared to the direct method and the multigrid method, this scheme needs substantially more computing time. It should only be used for test runs, e.g. to compare results with the other pressure solver methods.
-
pt_damping_factor#
- Fortran Type: R
-
Default: 0.0
Factor for damping the 3d-field of potential temperature.
In case of non-cyclic lateral boundary conditions (see bc_lr or bc_ns, a damping is applied to the potential temperature if a non-zero value is assigned to pt_damping_factor.
If switched on, the temperature is forced towards the value of its basic state (defined by the initial profile of the temperature). The intensity of damping is controlled by the value of pt_damping_factor. The damping starts weakly at a distance from the inflow boundary defined by pt_damping_width and rises according to a sin2-function to its maximum value at the inflow.
This method effectively damps gravity waves at the inflow boundary in case of non-cyclic lateral boundary conditions (see bc_lr or bc_ns). If the damping factor is too weak, gravity waves can still emerge within the damping area. If the damping factor is too strong, gravity waves may develop directly downstream of the damping area.
Note that the method has no effect if turbulent_inflow_mehod = 'absolute_value' or turbulent_inflow_mehod = 'absolute_value_thermodynamic'.
Detailed information about the damping can be found in the documentation of the non-cyclic lateral boundary conditions.
pt_damping_width#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: m
Width of the damping area for the potential temperature.
In case of non-cyclic lateral boundary conditions (see bc_lr or bc_ns), this parameter determines the range where damping of the potential temperature is applied. The damping domain starts at the inflow boundary and ranges up to a distance given by the value of pt_damping_width. The intensity of the damping is given by pt_damping_factor. pt_damping_width must not be lesser than zero or greater than the model domain length in the respective direction.
pt_reference#
- Fortran Type: R
- Default: Value of pt_surface
-
SI-Unit: K
Reference temperature to be used in the buoyancy term.
This parameter only becomes effective if reference_state = 'single_value' has been chosen for the reference state to be used in the buoyancy term.
Attention: This parameter has no effect in case of the ocean mode switched on, where the in-situ density is used in the buoyancy term (see reference_state for more details).
pt_surface#
- Fortran Type: R
- Default: 300.0
-
SI-Unit: K
Surface potential temperature.
This parameter assigns the value of the potential temperature
ptat the surface (k=0). Starting from this value, the initial vertical temperature profile is constructed via pt_vertical_gradient and pt_vertical_gradient_level. This profile is also used for the 1d-model as a stationary condition.Attention: In case of ocean mode runs, this parameter gives the temperature value at the sea surface, which is defined to be at
k= nzt. The profile is then constructed from the surface down to the bottom of the model (sea floor).
pt_surface_heating_rate#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: K h-1
Linear rate of surface temperature increase.
Instead of prescribing a surface_heatflux also a surface heating rate can be set via pt_surface_heating_rate. Surface cooling can be achieved by assigning a negative value. Setting of bc_pt_b = 'dirichlet' is required. The surface heating rate can be combined with pt_surface_initial_change.
pt_surface_heating_rate is allowed to be set as runtime parameter as well. In case of a restart run, it then overwrites the setting from the previous run and the new value will be used for further restarts, as long as no new setting via the runtime parameter namelist is made.
pt_surface_initial_change#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: K
Change in surface temperature to be applied at the beginning of a 3d run.
If pt_surface_initial_change is set to a non-zero value, the near surface sensible heat flux is not allowed to be given simultaneously (see surface_heatflux).
pt_surface_initial_change is allowed to be set as runtime parameter as well. It will apply for the respective run only. If a temperature change as wanted at the beginning of the next restart run too, pt_surface_initial_change has to be given in the runtime parameter namelist again.
pt_vertical_gradient#
- Fortran Type: R (10)
- Default: 0.0
-
SI-Unit: K / 100 m
Temperature gradient(s) of the initial temperature profile.
This temperature gradient holds starting from the height level defined by pt_vertical_gradient_level (precisely: for all uv levels
kwherezu(k)> pt_vertical_gradient_level,pt_init(k)is set:pt_init(k) = pt_init(k-1) + dzu(k) *pt_vertical_gradient) up to the top boundary or up to the next height level defined by pt_vertical_gradient_level. Below the first prescribed height level, the temperature is implicitly set constant with height. A total of 10 different gradients for 11 height intervals (10 intervals if pt_vertical_gradient_level(1) = 0.0) can be assigned. The surface temperature is assigned via pt_surface.Example:
pt_vertical_gradient = 1.0, 0.5,
pt_vertical_gradient_level = 500.0, 1000.0,
defines the temperature profile to be neutrally stratified up to z = 500.0 m with a temperature given by pt_surface. For 500.0 m < z <= 1000.0 m the temperature gradient is 1.0 K / 100 m and for z > 1000.0 m up to the top boundary it is 0.5 K / 100 m (it is assumed here that the assigned height levels correspond with uv levels).Attention:
In case of ocean mode runs, the profile is constructed like described above, but starting from the sea surface (k=nzt) down to the bottom boundary of the model. Height levels have then to be given as negative values, e.g. pt_vertical_gradient_level = -500.0, -1000.0.
pt_vertical_gradient_level#
- Fortran Type: R (10)
- Default: undefined
-
SI-Unit: m
Height level above which the temperature gradient defined by pt_vertical_gradient is effective.
The height levels have to be assigned in ascending order. The default values result in a neutral stratification regardless of the values of pt_vertical_gradient (unless the top boundary of the model is higher than 100000.0 m). For the piecewise construction of temperature profiles see pt_vertical_gradient.
Attention:
In case of ocean mode runs, the (negative) height levels have to be assigned in descending order.
q_surface#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: kg kg-1
Surface water vapor / total water mixing ratio.
This parameter assigns the value of the mixing ratio
qat the surface (k=0). Starting from this value, the initial humidity profile is constructed with q_vertical_gradient and q_vertical_gradient_level. This profile is also used for the ../../tec/1dmodel 1d-model as a stationary profile.
q_surface_initial_change#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: kg kg-1
Change in surface water vapor / total water mixing ratio to be applied at the beginning of the 3d run.
If q_surface_initial_change is set to a non-zero value, the near surface latent heat flux (water flux) is not allowed to be given simultaneously (see surface_waterflux).
q_vertical_gradient#
- Fortran Type: R (10)
- Default: 0.0
-
SI-Unit: kg kg-1 / 100 m
Humidity gradient(s) of the initial humidity profile.
This humidity gradient holds starting from the height level defined by q_vertical_gradient_level (precisely: for all uv levels
kwherezu(k)> q_vertical_gradient_level,q_init(k)is set:q_init(k) = q_init(k-1) + dzu(k) *q_vertical_gradient) up to the top boundary or up to the next height level defined by q_vertical_gradient_level. A total of 10 different gradients for 11 height intervals (10 intervals if q_vertical_gradient_level(1) = 0.0) can be assigned. The surface humidity is assigned via q_surface.Example:
q_vertical_gradient = 0.001, 0.0005,
q_vertical_gradient_level = 500.0, 1000.0,
defines the humidity to be constant with height up to z = 500.0 m with a value given by q_surface. For 500.0 m < z <= 1000.0 m the humidity gradient is 0.001 / 100 m and for z > 1000.0 m up to the top boundary it is 0.0005 / 100 m (it is assumed here that the assigned height levels correspond with uv levels).
q_vertical_gradient_level#
- Fortran Type: R (10)
- Default: undefined
-
SI-Unit: m
Height level from which on the humidity gradient defined by q_vertical_gradient is effective.
The height levels are to be assigned in ascending order. The default results in a humidity constant with height regardless of the values of q_vertical_gradient (unless the top boundary of the model is higher than 100000.0 m). For the piecewise construction of humidity profiles see q_vertical_gradient.
random_generator#
- Fortran Type: C*20
-
Default: random-parallel
Random number generator to be used for creating uniformly distributed random numbers.
The random generator is mainly used if random perturbations are to be imposed on the velocity field or on the surface heat flux field (see create_disturbances and random_heatflux), but also for other purposes, e.g. to add random velocity fluctuations to Lagrangian particles, or for the synthetic inflow turbulence generator.
Note:
Results from two otherwise identical model runs will not compare one-to-one if they have used different random number generators, or if they are different members of an ensemble of runs (see parameter ensemble_member_nr).Currently 3 choices are available:
-
random-parallel
A fully parallelized random number generator, which creates random numbers in a reproducible order which is independent from the number of cores used for a specific run, i.e. independent from the domain decomposition. Runs with different domain decomposition but the same total domain should produce identical results. The parallel random number generator also provides ensemble run capability, which can be steered by specifying the parameter ensemble_member_nr.
-
numerical-recipes
Random number generator from the "Numerical Recipes" book. Results are independent from the domain decomposition only for the perturbations imposed on the velocity field (see create_disturbances) but otherwise not. CPU-time for this generator does not scale with the number of cores, so it may become a bottleneck if larger number of cores (>100) are used. random_generator = 'numerical-recipes' is not allowed if the synthetic turbulence generator module is used.
-
system-specific
The Fortran built-in random number generator
RANDOM_NUMBER. Results are independent from the domain decomposition only for the perturbations imposed on the velocity field (see create_disturbances) but otherwise not.random_generator = 'system-specific' is not allowed if the synthetic turbulence generator module is used.
-
random_heatflux#
- Fortran Type: L
-
Default: .FALSE.
Parameter to impose random perturbations to the prescribed heat flux at surface grid points (internally stored in 1d-array
surf%shf).If a near surface heat flux is used as a bottom boundary condition (see surface_heatflux), it is by default assumed to be horizontally homogeneous. Random perturbations can be imposed on all respective surface grid points by setting random_heatflux = .T.. The disturbed heat flux is calculated by multiplying the value as given by surface_heatflux at each respective grid point with a normally distributed random number with mean value and standard deviation of 1. This is repeated after every time step.
In case of a non-flat topography, random_heatflux = .T.. imposes random perturbations on the combined heat flux composed of surface_heatflux at the bottom surface and wall_heatflux(0) at the topography top face. Furthermore, perturbations are only added if the respective surface is upward facing.
rans_const_c#
- Fortran Type: R (5)
-
Default: [0.55, 1.44, 1.92, 1.44, 0.0]
Constants used in the RANS mode turbulence closure.
The internally used variable is dimensioned as
rans_const_c(0:4). Array elements refer to constants \(c_0 = \sqrt[4]{c_\mu}\), \(c_1\), \(c_2\), \(c_3\), \(c_4\), that are used in the RANS equations as follows:rans_const_c(0) = \(c_0\)
rans_const_c(1) = \(c_1\)
rans_const_c(2) = \(c_2\)
rans_const_c(3) = \(c_3\)
rans_const_c(4) = \(c_4\)The default values are according to the standard TKE-e turbulence closure (see turbulence_closure).
rans_const_sigma#
- Fortran Type: R (2)
-
Default: [1.0, 1.3]
Constants used in the RANS mode turbulence closure.
The internally used variable is dimensioned
rans_const_sigma(2)and respective constants \(\sigma_e\) and \(\sigma_\epsilon\) that are used in the turbulence closure refer to the array elements as follows:rans_const_sigma(1) = \(\sigma_e\)
rans_const_sigma(2) = \(\sigma_\epsilon\)The default values are according to the standard TKE-e turbulence closure (see turbulence_closure).
rayleigh_damping_factor#
- Fortran Type: R
-
Default: 0.0
Factor for Rayleigh damping.
A so-called Rayleigh damping is applied to all prognostic variables if a non-zero value is assigned to rayleigh_damping_factor. If switched on, horizontal velocities, temperature, humidity/scalar (if switched on) and salinity (in case of ocean mode) are forced towards the value of their respective basic states (defined by the initial profiles of the geostrophic wind, temperature, etc., or in case of offline nesting, defined by the current mean of wind components, temperature, etc. of the mesoscale model). In case of large-scale subsidence (see subs_vertical_gradient) the basic state of temperature and humidity is adjusted with respect to the subsidence. Scalar quantities can be excluded from the damping (see scalar_rayleigh_damping). The intensity of damping is controlled by the value the rayleigh_damping_factor is assigned to. The damping starts weakly at a height defined by rayleigh_damping_height and rises according to a sin2-function to its maximum value at the top (ocean: bottom) boundary.
This method effectively damps gravity waves, caused by boundary layer convection, which may spread out vertically in the inversion layer and which are reflected at the top (ocean: bottom) boundary.
The Rayleigh damping factor must hold the condition 0.0 <= rayleigh_damping_factor <= 1.0. Large values (close to 1.0) may cause numerical instabilities.
rayleigh_damping_height#
- Fortran Type: R
- Default: Depends on atmosphere or ocean mode
-
SI-Unit: m
Height above (in ocean below) which the Rayleigh damping starts.
With Rayleigh damping switched on (see rayleigh_damping_factor), this parameter determines the range where damping is applied. By default, Rayleigh damping will be applied in the upper (ocean: lower) third of the model domain. Exactly rayleigh_damping_height = 2/3 *
zu(nzt)in the atmosphere, and rayleigh_damping_height = 2/3 *zu(nzb)in the ocean.
reference_state#
- Fortran Type: C*20
-
Default: initial_profile
This parameter defines the reference state that is used in the buoyancy terms.
Currently 3 choices are available:
-
initial_profile
The initial vertical potential temperature profile will be used. See pt_surface and pt_vertical_gradient for how to set the initial profile.
In case of runs with humidity, the virtual potential temperature will be used instead. See q_surface and q_vertical_gradient for how to set the initial water vapor mixing ratio profile.
In ocean mode runs, the in-situ density is used instead of temperature. In-situ density is calculated from the initial potential temperature and salinity profile. See sa_surface and sa_vertical_gradient for how to set the initial salinity profile.
In case of initializing_actions= * 'cyclic_fill'*, the main run uses the initial profile of the precursor run.
-
horizontal_average
The instantaneous horizontally averaged potential temperature profile will be used. Please be aware that this causes the reference state to change in time. This will increase the mean (horizontally averaged) buoyancy which then triggers a mean vertical velocity and in turn the divergence in the preliminary velocity field will increase in time, but the pressure solver will take care of it and removes any mean vertical velocities.
In case of runs with humidity, the virtual potential temperature will be used instead. In ocean mode runs, in-situ density is used.
-
single_value
A constant single potential temperature value is used as reference state. The respective value can be defined with parameter pt_reference.
Warning:
In case of runs with humidity, the virtual potential temperature is used. The reference value is then calculated from pt_reference and the surface water vapor mixing ratio q_surface, i.e. it cannot be explicitly set by the user.In ocean mode runs, the reference value cannot be explicitly set by the user. Instead, it is calculated as the vertical average of the initial potential density profile.
-
residual_limit#
- Fortran Type: R
- Default: 0.0001
-
SI-Unit: s-2m-3
Largest residual permitted for the multigrid scheme.
This is a parameter to steer the accuracy of the multigrid scheme (see psolver = 'multigrid'). The divergence of the 3D velocity field will be reduced iteratively as long as the calculated residual is greater than residual_limit. After a maximum number of 1000 iterative cycles, PALM aborts with a corresponding error message.
The reciprocal value of this parameter can be interpreted as a factor by which the divergence of the provisional velocity field is approximately reduced after the multigrid scheme has been applied (thus, the default value causes a reduction of the absolute value of divergence by approx. 6 orders of magnitude).
restart_data_format#
- Fortran Type: C*20
-
Default: mpi_shared_memory
Binary format of the input and output restart files.
This parameter can also be used in the
&runtime_parametersnamelist. Parameters restart_data_format_input and restart_data_format_output allow different formats to be used for input and output.Currently 3 choices are available:
-
fortran_binary
Each core (MPI thread) reads/writes its own file in Fortran binary format. I/O transfer rate is highest with this setting, but the large number of files that are generated if many cores are used may sometimes cause problems with parallel file systems (especially the Lustre file system, where hanging situations may appear).
-
mpi
I/O of all cores is directed to/from a single file using MPI-I/O. This method can also be used in serial mode (when PALM has been compiled without the
-D__paralleloption). In such a case, restart I/O is carried out using POSIX calls. MPI-I/O shows transfer rates that are a bit smaller than those for Fortran I/O. -
mpi_shared_memory
With this setting, I/O is performed only by a limited number of cores on each of the compute nodes a run is using. With restart_data_format = 'mpi' each core (MPI thread) writes and reads its relevant data. With restart_data_format = 'mpi_shared_memory', only every 4th (or any other multiple integer of the cores per node) reads and writes data and distributes it to the other cores. This is possible since all processes on a node can share their memory. With this, the I/O rates may increase especially for many-core CPUs.
Attention:
I/O-performance can often be further improved via specific file system settings, e.g. the number of I/O streams assigned to a respective file.
-
restart_data_format_input#
- Fortran Type: C*20
-
Default: Value of restart_data_format
Binary format of the input restart file.
See restart_data_format for allowed values. This parameter can also be used in the
&runtime_parametersnamelist.
restart_data_format_output#
- Fortran Type: C*20
-
Default: Value of restart_data_format
Binary format of the output restart file.
See restart_data_format for allowed values. This parameter can also be used in the
&runtime_parametersnamelist.
rotation_angle#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: degree
Rotation angle of the model's North direction relative to geographical North (clockwise rotation).
The rotation angle affects the Coriolis force as well as the solar radiation wihin the model domain. Further, it is used for correct geo-referencing each model grid point to real-world coordinates.
If a static driver is used, this parameter will be overwritten by the value given in the static driver.
roughness_length#
- Fortran Type: R
- Default: 0.1
-
SI-Unit: m
Roughness length assumed for the surface layer.
The roughness_length has to be smaller than the height of the constant flux layer that is used, which is defined by
dz/2.The roughness length for scalars can be assigned a value different from the one for momentum (see z0h_factor).
This parameter is effective only in case that a constant flux layer is switched on at the bottom boundary (see constant_flux_layer).
s_surface#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: kg m-3 (particles) or ppm (gases)
Surface concentration of the passive scalar.
This parameter assigns the value of the passive scalar
sat the surface (k=0). Starting from this value, the initial vertical scalar concentration profile is constructed using s_vertical_gradient and s_vertical_gradient_level.
s_surface_initial_change#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: kg m-3 (particles) or ppm (gases)
Change in surface scalar concentration to be applied at the beginning of the 3d run.
If s_surface_initial_change is set to a non-zero value, the near surface scalar flux is not allowed to be given simultaneously (see surface_scalarflux).
s_vertical_gradient#
- Fortran Type: R (10)
- Default: 0.0
-
SI-Unit: kg m-3 / 100 m (particles) or ppm / 100m (gases)
Scalar concentration gradient(s) of the initial scalar concentration profile.
The scalar gradient holds starting from the height level defined by s_vertical_gradient_level (precisely: for all uv levels
kwherezu(k)> s_vertical_gradient_level,s_init(k)is set:s_init(k) = s_init(k-1) + dzu(k) *s_vertical_gradient) up to the top boundary or up to the next height level defined by s_vertical_gradient_level. A total of 10 different gradients for 11 height intervals (10 intervals if s_vertical_gradient_level(1) = 0.0) can be assigned. The surface scalar value is assigned via s_surface.Example:
s_vertical_gradient = 0.1, 0.05,
s_vertical_gradient_level = 500.0, 1000.0,
defines the scalar concentration to be constant with height up to z = 500.0 m with a value given by s_surface. For 500.0 m < z <= 1000.0 m the scalar gradient is 0.1 kg m-3 / 100 m and for z > 1000.0 m up to the top boundary it is 0.05 kg m-3 / 100 m (it is assumed here that the assigned height levels correspond with uv levels).
s_vertical_gradient_level#
- Fortran Type: R (10)
- Default: undefined
-
SI-Unit: m
Height level from which on the scalar gradient defined by s_vertical_gradient is effective.
The height levels are to be assigned in ascending order. The default values result in a scalar concentration constant with height regardless of the values of s_vertical_gradient (unless the top boundary of the model is higher than 100000.0 m). For the piecewise construction of scalar concentration profiles see s_vertical_gradient.
scalar_advec#
- Fortran Type: C*16
-
Default: ws-scheme
Advection scheme to be used for the scalar quantities.
Currently 3 choices are available:
-
ws-scheme
The 5th order upwind scheme of Wicker and Skamarock (2002, Mon. Wea. Rev, 130, 2088-2097). The 5th order scheme implies a small numerical dissipation that stabilizes the solution. To assure a stable numerical solution, timestep_scheme = 'runge-kutta-3' has to be used. The Wicker-Skamarock scheme is based on on the advection term given in flux form, which requires the flow to be free of divergence, else a stable numerical solution is not guaranteed. Therefore, call_psolver_at_all_substeps = .T. should be set. The dispersion of this scheme error is much smaller than the dispersion error of the 'pw-scheme'.
The calculation of turbulent fluxes takes place inside the advection routines to get results that are consistent with the numerical solution.
Note: Due to the larger numerical stencil of this scheme, vertical grid stretching should be handled with care.
-
pw-scheme
The scheme of Piacsek and Williams (1970, J. Comp. Phys., 6, 392-405) with central finite differences in the form C3 is used.
Note: Compared with the 'ws-scheme', this scheme usually shows much stronger numerical dispersion. It is not recommended to be used in general.
-
bc-scheme
The Bott scheme modified by Chlond (1994, Mon. Wea. Rev., 122, 111-125). This is a conservative monotonous scheme with very small numerical diffusion and therefore very good conservation of scalar flow features. The scheme, however, is computationally very expensive, both because it is expensive itself and because it does (so far) not allow specific code optimizations (e.g. cache optimization). Choice of this scheme forces the Euler time step scheme to be used for the scalar quantities. For output of horizontally averaged profiles of the resolved / total heat flux, data_output_pr = 'w*theta*BC' / 'wthetaBC' should be used, instead of the standard profiles ('w*theta'* and 'wtheta') because these are too inaccurate with this scheme. However, for subdomain analysis (see statistic_regions) exactly the reverse holds: here 'w*theta*BC' and 'wthetaBC' show very large errors and should not be used.
This scheme is not allowed for non-cyclic lateral boundary conditions (see bc_lr and bc_nc) and requires loop_optimization = 'vector'.
-
scalar_rayleigh_damping#
- Fortran Type: L
-
Default: .TRUE.
Application of Rayleigh damping to scalars.
With Rayleigh damping switched on (see rayleigh_damping_factor), this parameter determines if the damping is applied also to scalars (temperature, humidity/scalar, salinity) or to the horizontal velocity components only.
spinup_time#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: s
Parameter for steering the length of the soil/wall spinup phase.
The soil/wall spinup allows for the adjustment of the inert soil- and wall-layer temperatures to the prevailing atmospheric conditions, prior to the actual 3D atmosphere simulation. This saves a significant amount of computational time because during the spinup phase, the soil and wall models run without an interactive atmosphere.
Currently, this mechanism is implemented for the land surface and the urban surface model. In the spinup phase, these models predict the skin and soil/wall temperatures. All window temperatures (surface and deeper layers) are not calculated during spinup. For using the spinup mechanism it is required to have a radiation model activated. Forcing is achieved by radiation and optionally a varying near-surface air temperature (steered via spinup_pt_mean, spinup_pt_amplitude, and the incoming solar radiation. After completion of the spinup, the atmospheric code is switched on and runs coupled to the surface models. On the timeline, the spinup phase has negative times counting towards 0 s, which is when the atmosphere is activated.
Note that the near-surface temperatures are reset after the spinup to their initial values prescribed by the user. The spinup output can be steered via the additional parameters dt_run_control_spinup and data_output_during_spinup.
Note that it is not allowed to set spinup_time for childs that have been activated in a restart run for the first time.
spinup_pt_amplitude#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: K
Representative amplitude of the diurnal near-surface temperature variation during the spinup phase (see spinup_time).
Note that this amplitude does not match the true temperature amplitude during spinup. The true amplitude depends also on the incoming shortwave radiation and thus depends on the day of the year and the geographic location.
spinup_pt_mean#
- Fortran Type: R
- Default: Value of pt_surface
-
SI-Unit: K
Mean near-surface air temperature during the spinup phase (see spinup_time).
If no value is provided, the mean will be inferred from the setting of pt_surface.
statistic_regions#
- Fortran Type: I
-
Default: 0
Number of additional user-defined subdomains for which statistical analysis and corresponding output (profiles, time series) shall be made.
By default, vertical profiles and other statistical quantities are calculated as horizontal and/or volume average of the total model domain. Beyond that, these calculations can also be carried out for subdomains which can be defined using the array
rmaskwithin the user-defined software (see user-defined domains). The number of these subdomains is determined with the parameter statistic_regions. Maximum 9 additional subdomains are allowed. The parameter region can be used to assign names (identifier) to these subdomains which are then used in the headers of the output files and plots.Data for the total domain and all defined subdomains are output to the same file(s) (DATA_1D_PR_NETCDF, DATA_1D_TS_NETCDF). In case of statistic_regions > 0, data for the different domains can be distinguished by a suffix which is appended to the quantity names. Suffix 0 means data for the total domain, suffix 1 means data for subdomain 1, etc.
subs_vertical_gradient#
- Fortran Type: R (10)
- Default: 0.0
-
SI-Unit: m s-1 / 100 m
Vertical gradient(s) of the profile for the large-scale subsidence/ascent velocity.
This gradient holds starting from the height level defined by subs_vertical_gradient_level (precisely: for all uv levels
kwherezu(k)> subs_vertical_gradient_level,w_subs(k)is set:w_subs(k) = w_subs(k-1) + dzu(k) *subs_vertical_gradient) up to the top boundary or up to the next height level defined by subs_vertical_gradient_level. A total of 10 different gradients for 11 height intervals (10 intervals if subs_vertical_gradient_level(1) = 0.0) can be assigned.Example:
subs_vertical_gradient = -0.002, 0.0,
subs_vertical_gradient_level = 0.0, 1000.0defines the subsidence/ascent profile to be linear up to z = 1000.0 m with a surface value of 0 m/s. Due to the gradient of -0.002 m s-1 / 100 m the subsidence velocity has a value of -0.02 m s-1 in z = 1000.0 m. For z > 1000.0 m up to the top boundary the gradient is 0.0 m s-1 / 100 m (it is assumed that the assigned height levels correspond with uv levels). This results in a subsidence velocity of -0.02 m s-1 at the top boundary.
With an appropriate construction of
w_subs, the boundary layer height \(z_i\) can be kept approximately constant.
subs_vertical_gradient_level#
- Fortran Type: R (10)
- Default: 0.0
-
SI-Unit: m
Height level from which on the gradient for the subsidence/ascent velocity defined by subs_vertical_gradient is effective.
The height levels have to be assigned in ascending order. The default values result in a profile which is zero everywhere regardless of the values of subs_vertical_gradient. For the piecewise construction of the subsidence/ascent velocity profile see subs_vertical_gradient.
surface_heatflux#
- Fortran Type: R
- Default: undefined
-
SI-Unit: depends on 'flux_input_mode'
Sensible heat flux at the bottom surface.
If a value is assigned to this parameter, the internal surface heat flux array
surf%shfis initialized with the value of surface_heatflux as the bottom (horizontally homogeneous) boundary condition for the temperature equation. This additionally requires that a Neumann condition must be used for the potential temperature (see bc_pt_b), because otherwise the resolved scale may contribute to the surface flux so that a constant value cannot be guaranteed. Also, changes of the surface temperature (see pt_surface_initial_change) are not allowed when surface_heatflux is given. The parameter random_heatflux can be used to impose random perturbations on the (homogeneous) surface heat flux.In case of a non-flat topography, the internal surface heat flux
surf%shfis initialized with the value of surface_heatflux at the bottom surface and wall_heatflux(0) at the topography top face. The parameter random_heatflux can be used to impose random perturbations on this combined surface heat flux.If no surface heat flux is assigned, it is calculated at each time step by \(u_* \theta_*\) (when constant_flux_layer is switched on). Here, \(u_*\) and \(\theta_*\) are calculated from Monin-Obukhov similarity theory assuming logarithmic wind and temperature profiles between k=0 and k=1. In this case, a Dirichlet condition (see bc_pt_b must be used as bottom boundary condition for the potential temperature.
Non-zero values must not be given for surface_heatflux in case of simulations with pure neutral stratification (see parameter neutral).
surface_heatflux is allowed to be set as runtime parameter as well. In case of a restart run, it then overwrites the setting from the previous run and the new value will be used for further restarts, as long as no new setting via the runtime parameter namelist is made.
See also top_heatflux.
Note: Depending on flux_input_mode, the value of surface_heatflux has to be given either in unit K ms-1 (kinematic flux), or in W m-2.
Attention: If the constant flux layer has been switched off (constant_flux_layer = .F.), setting of surface_heatflux requires setting of use_surface_fluxes = .T..
surface_pressure#
- Fortran Type: R
- Default: 1013.25
-
SI-Unit: hPa
Atmospheric pressure at the surface.
Starting from this surface value, the vertical pressure profile is calculated once at the beginning of the run assuming a neutrally stratified atmosphere. This is needed for converting between the liquid water potential temperature and the potential temperature (see bulk_cloud_model = .T.). Note, if initializing_actions = 'read_from_file', surface_pressure will be overwritten by the value given in the dynamic input file.
surface_scalarflux#
- Fortran Type: R
- Default: undefined
-
SI-Unit: kg m-2 s-1 (particle flux) or ppm m s-1 (gaseous flux)
Scalar flux at the surface.
If a value is assigned to this parameter, the respective scalar flux value is used as bottom (horizontally homogeneous) boundary condition for the scalar concentration equation. This additionally requires that a Neumann condition must be used for the scalar concentration (see bc_s_b), because otherwise the resolved scale may contribute to the surface flux so that a constant value cannot be guaranteed. Also, changes of the surface scalar concentration (see s_surface_initial_change) are not allowed.
If no surface scalar flux is assigned, it is calculated at each time step by \(u_* s_*\) (of course only with constant_flux_layer switched on). Here, \(s_*\) is calculated from Monin-Obukhov similarity theory assuming a logarithmic scalar concentration profile between k=0 and k=1. In this case, a Dirichlet condition (see bc_s_b) must be used as bottom boundary condition for the scalar concentration.
surface_waterflux#
- Fortran Type: R
- Default: undefined
-
SI-Unit: kg kg-1 m s-1
Kinematic water flux near the surface.
If a non-zero value is assigned to this parameter, the respective water flux value is used as bottom (horizontally homogeneous) boundary condition for the humidity equation. This additionally requires that a Neumann condition must be used for the water vapor / total water mixing ratio (see bc_q_b), because otherwise the resolved scale may contribute to the surface flux so that a constant value cannot be guaranteed. Also, changes of the surface humidity (see q_surface_initial_change) are not allowed.
If no surface water flux is assigned, it is calculated at each time step by \(u_* q_*\) (of course only with a constant flux layer switched on). Here, \(q_*\) is calculated from Monin-Obukhov similarity theory assuming a logarithmic temperature profile between k=0 and k=1. In this case, a Dirichlet condition (see bc_q_b) must be used as the bottom boundary condition for the humidity.
terrain_following_mapping#
- Fortran Type: L
-
Default: .FALSE.
Parameter to enable simulations featuring terrain-following mapping of profile data in combination with cyclic-fill initialization.
If the simulation is initialized using the cyclic fill method (see initializing_actions = 'cyclic_fill' ), terrain_following_mapping = .T. shifts the initial 3d-arrays vertically depending on the local surface height of each horizontal grid point, i.e. the initial 3d-arrays are mapped in a terrain-following way.
Furthermore, in case of terrain_following_mapping = .T. the mean inflow profiles as used by the turbulence recycling method (see turbulent_inflow_method) are also shifted vertically with respect to the topography height at the inflow / recycling plane. For this, the topography height must be constant along the entire inflow / recycling plane.
terrain_following_mapping = .T. does not allow to use vertical grid stretching. Furthermore, the topography mode must be set to 'read_from_file' (see topography).
timestep_scheme#
- Fortran Type: C*20
-
Default: runge-kutta-3
Time step scheme to be used for the integration of the prognostic variables.
Independent from timestep_scheme a different time step scheme can be chosen for the subgrid-scale TKE using parameter use_upstream_for_tke.
Currently 3 choices are available:
-
runge-kutta-3
Third order Runge-Kutta scheme.
For further information see here. -
runge-kutta-2
Second order Runge-Kutta scheme.
Not allowed for momentum_advec = 'ws-scheme' or scalar_advec = 'ws-scheme'. For further information see here. -
euler
First order Euler scheme.
Not allowed for momentum_advec = 'ws-scheme' or scalar_advec = 'ws-scheme'.
-
topography#
- Fortran Type: C*40
-
Default: flat
Topography mode.
Alternatively to the available Topography modes explained below, the user may add code to the user interface subroutine user_init_grid to allow further topography modes. These require to explicitly set the topography_grid_convention to either 'cell_edge' or 'cell_center' .
Non-flat topography modes allow to assign a sensible wall_heatflux, a wall_humidityflux (requires humidity = .T.), or a wall_scalarflux (requires passive_scalar = .T.) at the five topography faces using the respective parameters.
All non-flat topography modes require the use of psolver /= 'sor' , alpha_surface = 0.0, galilei_transformation = .F., cloud_droplets = .F. (has not been tested), and constant_flux_layer = .T. (except closed channel flow).
Note:
The given topography is internally filtered so that surfaces with a distance of only one grid point (e.g. small street canyons) are filled up to the minimum topography height of the directly adjoining grid points along the x- and the y-direction. This is necessary, because for such poorly resolved features, the continuity equation is not fulfilled on a discrete grid, as the degree of freedom for the pressure solver is insufficient. Suchholesare suspected to lead to unrealistic velocity blow-ups, hence, they are filled up.Note that an inclined model domain requires the use of topography = 'flat' and a nonzero alpha_surface.
Currently 6 choices are available:
-
flat
Flat surface. The surface is assumed to be at grid index
k= 0. -
single_building
Flow around a single rectangular building mounted on an otherwise flat surface. The building size and location can be specified by the parameters building_height, building_length_x, building_length_y, building_wall_left and building_wall_south.
-
single_street_canyon
Flow over a single, quasi-2D street canyon of infinite length oriented either along x- or along y-direction. The canyon size, orientation, and location can be specified by the parameters canyon_height plus either canyon_width_x and canyon_wall_left or canyon_width_y and canyon_wall_south.
-
read_from_file
Arbitrary topography. This mode requires the input file TOPOGRAPHY_DATA, which contains the arbitrary topography height at each horizontal grid point in m (LOD2), or information about which of the model grid points is filled with topography/buildings (LOD3). These data must exactly match PALM's grid, except topography heights (LOD2), which are rounded to the nearest vertical PALM grid level.
-
tunnel
Flow within a tunnel of finite or infinite length oriented either along x- or along y-direction. The height of the tunnel outer top wall can be specified by tunnel_height. The tunnel length and width can be specified via tunnel_length and either tunnel_width_x or tunnel_width_y. Furthermore, the depth of tunnel walls can be specified by tunnel_wall_depth. Note that tunnels are always centered in the model domain along x- or y-direction.
-
closed_channel
Flow within a closed channel with walls at both the lower boundary at
k=0(forwand scalars) and the upper boundary atk=nzt(foww) andk=nzt+1(for scalars). Note that PALM's default grid structure (see spatial discretization) is modified in a closed channel so thatw(k=nzt)andscalar(k=nzt+1)are defined at the same height, because investigating such a flow requires a symmetric model domain along z. 'closed_channel' requires constant_flux_layer = .T.. and momentum_advec = scalar_advec = 'ws-scheme'. Grid stretching along z and ocean mode are not allowed.
-
topography_grid_convention#
- Fortran Type: C*11
-
Default: Depends on value of topography
Convention for defining the topography grid.
This parameter only needs to be used for topography /= 'read_from_file'. The default value is 'cell_edge', if topography = 'single_building' or 'single_street_canyon', and 'cell_center' if topography = 'read_from_file', or 'tunnel'. For simulation setups using a user-defined topography, topography_grid_convention must be explicitly set to either 'cell_edge' or 'cell_center'.
Currently 2 choices are available:
-
cell_edge
The distance between cell edges defines the extent of topography. This setting is normally used for generic topographies, i.e. topographies that are constructed using length parameters. For example, topography = 'single_building' is constructed using building_length_x and building_length_y. The advantage of this setting is that the actual size of generic topography is independent of the grid size, provided that the length parameters are an integer multiple of the grid lengths dx and dy. This is convenient for resolution parameter studies.
-
cell_center
The number of topography cells defines the extent of topography. This setting is normally for rastered real topographies derived from digital elevation models. For example, topography = 'read_from_file' is constructed using the input file TOPOGRAPHY_DATA. The advantage of this setting is that the rastered topography cells of the input file are directly mapped to PALM's topography grid boxes.
-
top_heatflux#
- Fortran Type: R
- Default: undefined
-
SI-Unit: depends on 'flux_input_mode'
Kinematic sensible heat flux at the top boundary.
If a value is assigned to this parameter, the top boundary sensible heat flux is initialized with the value of top_heatflux as the top (horizontally homogeneous) boundary condition for the temperature equation. This additionally requires that a Neumann condition must be used for the potential temperature (see bc_pt_t), because otherwise the resolved scale may contribute to the top flux so that a constant flux value cannot be guaranteed.
The application of a top heat flux additionally requires the setting of initial parameter use_top_fluxes = .T..
No constant flux layer is available at the top boundary so far.
Non-zero values must not be given for top_heatflux in case of simulations with pure neutral stratification (see parameter neutral.
See also surface_heatflux.
Note: Depending on flux_input_mode, the value of top_heatflux has to be given either in unit K ms-1 (kinematic flux), or in W m-2.
top_momentumflux_u#
- Fortran Type: R
- Default: undefined
-
SI-Unit: m2 s-2
Vertical momentum flux of
uat the top boundary.The respective grid points at the top boundary are initialized with the value of top_momentumflux_u as the top (horizontally homogeneous) boundary condition for the u-momentum equation. Data is stored within the internal surface data array
surf%usws.The application of a top momentum flux additionally requires the setting of initialization parameter use_top_fluxes = .T.. Setting of top_momentumflux_u requires setting of top_momentumflux_v, too.
A Neumann condition should be used for the
uvelocity component (see bc_uv_t), because otherwise the resolved scale may contribute to the top flux so that a constant flux value cannot be guaranteed.No constant flux layer is available at the top boundary so far.
The coupled ocean parameter file PARIN_O should include dummy assignments to both top_momentumflux_u and top_momentumflux_v (e.g. top_momentumflux_u = 0.0, top_momentumflux_v = 0.0) to enable the momentum flux coupling.
top_momentumflux_v#
- Fortran Type: R
- Default: undefined
-
SI-Unit: m2 s-2
Vertical momentum flux of
vat the top boundary.The respective grid points at the top boundary are initialized with the value of top_momentumflux_v as the top (horizontally homogeneous) boundary condition for the v-momentum equation. Data is stored within the internal surface data array
surf%vsws.The application of a top momentum flux additionally requires the setting of initialization parameter use_top_fluxes = .T.. Setting of top_momentumflux_v requires setting of top_momentumflux_u, too.
A Neumann condition should be used for the
vvelocity component (see bc_uv_t), because otherwise the resolved scale may contribute to the top flux so that a constant flux value cannot be guaranteed.No constant flux layer is available at the top boundary so far.
The coupled ocean parameter file PARIN_O should include dummy assignments to both top_momentumflux_u and top_momentumflux_v (e.g. top_momentumflux_u = 0.0, top_momentumflux_v = 0.0) to enable the momentum flux coupling.
top_scalarflux#
- Fortran Type: R
- Default: undefined
-
SI-Unit: kg m-2 s-1 (particle flux) or ppm m s-1 (gaseous flux)
Vertical scalar flux at the top boundary.
The respective grid points at the top boundary are initialized with the value of top_scalarflux as the top (horizontally homogeneous) boundary condition for the scalar equation. Data is stored within the internal surface data array
surf%ssws.The application of a top scalar flux additionally requires the setting of initialization parameter use_top_fluxes = .T..
A Neumann condition should be used for the scalar (see bc_s_t), because otherwise the resolved scale may contribute to the top flux so that a constant flux value cannot be guaranteed.
No constant flux layer is available at the top boundary so far.
tunnel_height#
- Fortran Type: R
- Default: Value of zw(INT(0.2*nz))
-
SI-Unit: m
Height of the tunnel (outer wall).
Parameter comes into effect only in case of topography = 'tunnel'.
tunnel_length#
- Fortran Type: R
- Default: 9999999.9
-
SI-Unit: m
Length of the tunnel.
Parameter comes into effect only in case of topography = 'tunnel'.
tunnel_length can be smaller (open tunnel) or larger (infinite tunnel) than the extension of the model domain along the tunnel axis.
tunnel_wall_depth#
- Fortran Type: R
- Default: Value of MAX(dx, dy, dz)
-
SI-Unit: m
Depth of the tunnel walls.
Parameter comes into effect only in case of topography = 'tunnel'.
tunnel_width_x#
- Fortran Type: R
- Default: undefined
-
SI-Unit: m
Width of the tunnel along x-direction with respect to the tunnel outer walls.
Parameter comes into effect only in case of topography = 'tunnel'.
If parameter is set, it indicates a tunnel orientation along
x. The tunnel width must be at least larger than 2 * dx + 2 * tunnel_wall_depth.
tunnel_width_y#
- Fortran Type: R
- Default: 9999999.9
-
SI-Unit: m
Width of the tunnel along y-direction with respect to the tunnel outer walls.
Parameter comes into effect only in case of topography = 'tunnel'.
If parameter is set, it indicates a tunnel orientation along
y. The tunnel width must be at least larger than 2 * dy + 2 * tunnel_wall_depth.
turbulence_closure#
- Fortran Type: C*20
-
Default: 1.5-order
Parameter to choose between different turbulence closures.
Currently 5 choices are available:
-
1.5-order
Turbulence closure according to a modified version of Deardorff`s subgrid-scale model (Deardorff 1980, Moeng & Wyngaard, 1988) as described by Saiki et al. (2000). See details in the documentation.
-
1.5-order-dai
Turbulence closure according to a modified version of Deardorffs subgrid-scale model described by Dai et al. (2020). The implementation is still not thoroughly tested and should be used with extra care. See details in the documentation.
-
dynamic
Use a dynamic subgrid-scale model according to Germano et al. (1991) and Heinz (2008). See details in the documentation.
-
tke-l
Turbulence closure utilising the Prandtl-Kolmogorov relation. See details in the documentation.
This option puts PALM into RANS mode.
-
tke-e
Standard TKE-e closure for Reynolds-averaged Navier-Stokes equations. The used constants can be changed using parameters rans_const_c and rans_const_sigma. See details in the documentation.
This option puts PALM into RANS mode.
-
turbulent_outflow#
- Fortran Type: L
-
Default: .FALSE.
Parameter to switch on a turbulent outflow boundary condition.
This outflow condition works technically similar to the turbulent recycling method at the inflow, but always instantaneous (not turbulent fluctuations) values of
u,v,w,pt,q,s, andeare copied from a vertical source plane (see outflow_source_plane) from inside the domain to the outflow boundary. This prevents numerical errors which can occur if negative velocity values appear at (and normal to) the outflow boundary while using the radiation boundary condition.For using this outflow method bc_lr = 'dirichlet/radiation' is required. It can only be used for an outflow boundary at the right model boundary, so far.
The vertical source plane should be placed not too close to the outflow boundary. This method replaces the radiation boundary condition at the outflow.
u_bulk#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: m s-1
u-component of predefined bulk velocity.
This parameter comes into effect if conserve_volume_flow = .T. and conserve_volume_flow_mode = 'bulk_velocity'.
u_profile#
- Fortran Type: R (200)
- Default: undefined
-
SI-Unit: ms-1
Values of u-velocity component to be used as initial profile.
The corresponding height levels have to be provided by parameter uv_heights. You need to give values for each of the height levels that are defined via uv_heights. The first velocity value always has to be assigned to the surface and must be zero, i.e. u_profile(1) = 0.0. Velocity values at the model grid levels are calculated from the given values/heights by linear interpolation. If the uppermost value is given for a height smaller than the domain height, this value will be used for all grid points above this level. In runs with non-cyclic horizontal boundary conditions this profile will be used as fixed mean inflow profile.
Note: Simultaneous use of forcing by geostrophic wind is not allowed, since this could lead to inconsistencies in the flow forcing. Hence, omega = 0.0 must be set when using u_profile and v_profile. The latter flow forcing is typically used for simulating wind-tunnel applications (where the effect of Coriolis force is negligible) in order to fairly reproduce the measured wind-tunnel profiles within the LES.
ug_surface#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: m s-1
u-component of the geostrophic wind at the surface.
This parameter assigns the value of the u-component of the geostrophic wind (
ug) at the surface (k=0). Starting from this value, the initial vertical profile of the u-component of the geostrophic wind is constructed with ug_vertical_gradient and ug_vertical_gradient_level. The profile constructed in that way is used for creating the vertical initial velocity profile of the 3d-model. Either it is applied, as it has been specified by the user (initializing_actions = 'set_constant_profiles' ) or it is used for calculating a stationary boundary layer wind profile (initializing_actions = 'set_1d-model_profiles' ).In case of a flat surface and cyclic horizontal boundary conditions, and if
ugis constant with height (i.e.ug(:)= ug_surface), one may use a Galilei-transformation of the coordinate system to allow for larger time steps (see galilei_transformation), in order to spare computational resources.Note: In case of ocean mode runs, this parameter gives the u-component of the geostrophic velocity value (i.e. the pressure gradient) at the sea surface, which is at
k=nzt. The profile is then constructed from the surface down to the bottom of the model.Attention: If the Coriolis force is switched off via omega = 0.0, ug_surface is still used for creating the initial wind profile, but of course it can't be called the profile of the geostrophic wind any more!
ug_vertical_gradient#
- Fortran Type: R (10)
- Default: 0.0
-
SI-Unit: m s-1 / 100 m
Vertical gradient(s) of the initial profile of the u-component of the geostrophic wind.
The gradient holds starting from the height level defined by ug_vertical_gradient_level (precisely: for all
uvlevelskwherezu(k)> ug_vertical_gradient_level,ug(k)is set:ug(k) = ug(k-1) + dzu(k) *ug_vertical_gradient) up to the top boundary or up to the next height level defined by ug_vertical_gradient_level. A total of 10 different gradients for 11 height intervals can be assigned (10 intervals if ug_vertical_gradient_level(1) = 0.0). The surface geostrophic wind is assigned by ug_surface.Note: In case of ocean mode runs, the profile is constructed like described above, but starting from the sea surface (
k=nzt) down to the bottom boundary of the model. Height levels have then to be given as negative values, e.g. ug_vertical_gradient_level = -500.0, -1000.0.
ug_vertical_gradient_level#
- Fortran Type: R (10)
- Default: 0.0
-
SI-Unit: m
Height level from which on the gradient defined by ug_vertical_gradient is effective.
The height levels have to be assigned in ascending order. For the piecewise construction of a profile of the u-component of the geostrophic wind \(u_g\) see ug_vertical_gradient.
Note: In case of ocean mode runs, the (negative) height levels have to be assigned in descending order.
use_fixed_date#
- Fortran Type: L
-
Default: .FALSE.
Switch to fix the calendar day within a simulation.
If use_fixed_date = .T., the calendar day does not change during a simulation. When the simulated time reaches 24:00:00 UTC, the time is reset to 00:00:00 UTC without the calendar day being changed. This affects, e.g., the solar position during the simulation.
Example: In case of origin_date_time = '2020-07-15 20:00:00 +00', the internal clock after 5h simulated time would be at '2020-07-15 01:00:00 +00'.
use_fixed_time#
- Fortran Type: L
-
Default: .FALSE.
Switch to fix the time of day within a simulation.
If use_fixed_time = .T., the time of day does not change during a simulation, i.e. the internal clock would always show the value set by origin_date_time. This results in the sun having a fixed position during the simulation.
use_free_convection_scaling#
- Fortran Type: L
-
Default: .FALSE.
Parameter to switch on the use of the local free convection velocity scale \(w_{lfc}\).
When switched on, \(w_{lfc}\) is added to the horizontal wind velocity for use in the constant flux layer parameterization to calculate the Obukhov length and the friction velocity \(u_*\) over horizontally-aligned surfaces. The horizontal velocity \(u_h\) at height of the first vertical grid level \(z_\mathrm{mo}\) is then calculated as:
\[u_\mathrm{h} = \left(u(z_\mathrm{mo})^2 + v(z_\mathrm{mo})^2 + w_\mathrm{lfc}^2\right)^{1/2}\]with
\[w_\mathrm{lfc} = \left( \frac{g}{\theta(z_\mathrm{mo})} * z_\mathrm{mo} * \overline{w'\theta'}_0\right)^{1/3}\]This is particularly useful in simulations of convective boundary layers where the local near-surface wind is expected to be close to zero, e.g. in urban environments when the present buildings create spots of stagnant air. \(w_{lfc}\) accounts for the dominant eddies close to the surface that cannot be resolved by the LES model and increases \(u_h\) by a small amount. When using the land surface scheme, this might prevent the surface sensible heat flux from dropping to zero under no-wind conditions.
use_ug_for_galilei_tr#
- Fortran Type: L
-
Default: .TRUE.
Switch to determine the translation velocity in case that a Galilean transformation is used.
In case of a Galilean transformation (see galilei_transformation), use_ug_for_galilei_tr = .T. ensures that the coordinate system is translated with the geostrophic wind speed.
Alternatively, use_ug_for_galilei_tr = .F. replaces the geostrophic wind as translation speed by the (volume) averaged velocity. However, in this case, fast growing gravity waves may develop, so this choice is usually not recommended.
use_subsidence_tendencies#
- Fortran Type: L
-
Default: .FALSE.
This parameter determines how the large-scale subsidence is computed for runs with large-scale forcing.
If use_subsidence_tendencies = .F., the large-scale subsidence velocity \(w_{subs}\) is used to advect the prognostic variables (subroutine
subsidence). If use_subsidence_tendencies = .T., the subroutinesubsidenceis not called and the subsidence tendencies from LSF_DATA are applied to the prognostic variables in the subroutinels_advec. The usage of use_subsidence_tendencies requires large_scale_forcing = .T. as well as large_scale_subsidence = .T..
use_surface_fluxes#
- Fortran Type: L
-
Default: .FALSE.
Parameter to steer the treatment of the subgrid-scale vertical fluxes within the diffusion terms at
k = 1(bottom boundary).By default, the near-surface subgrid-scale fluxes are parameterized (like in the remaining model domain) using a gradient approach. In case of use_surface_fluxes = .T., the SGS-fluxes are calculated via Monin-Obukhov similarity theory (depends on the bottom boundary conditions, see bc_pt_b, bc_q_b and bc_s_b), or user-assigned surface fluxes are used instead (see surface_heatflux, surface_waterflux and surface_scalarflux).
use_surface_fluxes is automatically set .T., if a constant flux layer is used (see constant_flux_layer).
use_top_fluxes#
- Fortran Type: L
-
Default: .FALSE.
Parameter to steer the treatment of the subgrid-scale vertical fluxes within the diffusion terms at
k = nz(top boundary).By default, the fluxes at grid level
nzare calculated using the gradient approach. If use_top_fluxes = .T., user-assigned fluxes are used instead (see top_heatflux, top_momentumflux_u, top_momentumflux_v, top_salinityflux).Currently, no value for the latent heatflux can be assigned. In case of use_top_fluxes = .T., the latent heat flux at the top will be automatically set to zero.
use_upstream_for_tke#
- Fortran Type: L
-
Default: .FALSE.
Parameter to choose the advection and time step scheme to be used for the subgrid-scale TKE.
By default, the advection scheme and the time step scheme to be used for the subgrid-scale TKE are set by the initialization parameters scalar_advec and timestep_scheme, respectively. use_upstream_for_tke = .T. forces the Euler-scheme and the upstream-scheme to be used as time step scheme and advection scheme. Using these schemes, strong artificial near-surface vertical gradients of the subgrid-scale TKE, as they are generated by some non-diffusive advection schemes, are avoided. use_upstream_for_tke = .T. is recommended when subgrid-scale velocities are considered for advection of particles (see use_sgs_for_particles) and an advection scheme for scalars scalar_advec /= 'ws-scheme' is used.
uv_heights#
- Fortran Type: R (200)
- Default: undefined
-
SI-Unit: m
Height levels in ascending order, for which prescribed u,v-velocities (u_profile, v_profile) are given.
The first height level must always be zero, i.e. uv_heights(1) = 0.0.
v_bulk#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: m s-1
v-component of predefined bulk velocity.
This parameter comes into effect if conserve_volume_flow = .T. and conserve_volume_flow_mode = 'bulk_velocity'.
v_profile#
- Fortran Type: R (200)
- Default: undefined
-
SI-Unit: m s-1
Values of v-velocity component to be used as initial profile.
The corresponding height levels have to be provided by parameter uv_heights. You need to give values for each of the height levels that are defined via uv_heights. The first velocity value always has to be assigned to the surface and must be zero, i.e. v_profile(1) = 0.0. Velocity values at the model grid levels are calculated from the given values/heights by linear interpolation. If the uppermost value is given for a height smaller than the domain height, this value will be used for all grid points above this level. In runs with non-cyclic horizontal boundary conditions this profile will be used as fixed mean inflow profile.
Note: Simultaneous use of forcing by geostrophic wind is not allowed, since this could lead to inconsistencies in the flow forcing. Hence, omega = 0.0 must be set when using u_profile and v_profile. The latter flow forcing is typically used for simulating wind-tunnel applications (where the effect of Coriolis force is negligible) in order to fairly reproduce the measured wind-tunnel profiles within the LES.
vdi_checks#
- Fortran Type: L
-
Default: .FALSE.
To switch on the internal controls according to VDI 3873 Part 9.
For more information see the PALM VDI evaluation report.
vg_surface#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: m s-1
v-component of the geostrophic wind at the surface.
This parameter assigns the value of the v-component of the geostrophic wind (
vg) at the surface (k=0). Starting from this value, the initial vertical profile of the v-component of the geostrophic wind is constructed with vg_vertical_gradient and vg_vertical_gradient_level. The profile constructed in that way is used for creating the vertical initial velocity profile of the 3d-model. Either it is applied, as it has been specified by the user (initializing_actions = 'set_constant_profiles' ) or it is used for calculating a stationary boundary layer wind profile (initializing_actions = 'set_1d-model_profiles' ).In case of a flat surface and cyclic horizontal boundary conditions, and if
vgis constant with height (i.e.vg(:)= vg_surface), one may use a Galilei-transformation of the coordinate system to allow for larger time steps (see galilei_transformation), in order to spare computational resources.Note: In case of ocean mode runs, this parameter gives the v-component of the geostrophic velocity value (i.e. the pressure gradient) at the sea surface, which is at
k=nzt. The profile is then constructed from the surface down to the bottom of the model.Attention: If the Coriolis force is switched off via omega = 0.0, vg_surface is still used for creating the initial wind profile, but of course it can't be called the profile of the geostrophic wind any more!
vg_vertical_gradient#
- Fortran Type: R (10)
- Default: 0.0
-
SI-Unit: m s-1 / 100 m
Vertical gradient(s) of the initial profile of the v-component of the geostrophic wind.
The gradient holds starting from the height level defined by vg_vertical_gradient_level (precisely: for all
uvlevelskwherezu(k)> vg_vertical_gradient_level,vg(k)is set:vg(k) = vg(k-1) + dzu(k) *vg_vertical_gradient) up to the top boundary or up to the next height level defined by vg_vertical_gradient_level. A total of 10 different gradients for 11 height intervals can be assigned (10 intervals if vg_vertical_gradient_level(1) = 0.0). The surface geostrophic wind is assigned by vg_surface.Note: In case of ocean mode runs, the profile is constructed like described above, but starting from the sea surface (
k=nzt) down to the bottom boundary of the model. Height levels have then to be given as negative values, e.g. vg_vertical_gradient_level = -500.0, -1000.0.
vg_vertical_gradient_level#
- Fortran Type: R (10)
- Default: 0.0
-
SI-Unit: m
Height level from which on the gradient defined by vg_vertical_gradient is effective.
The height levels have to be assigned in ascending order. For the piecewise construction of a profile of the v-component of the geostrophic wind \(v_g\) see vg_vertical_gradient.
Note: In case of ocean mode runs, the (negative) height levels have to be assigned in descending order.
wall_adjustment#
- Fortran Type: L
-
Default: .TRUE.
Parameter to restrict the mixing length in the vicinity of the bottom boundary as well as near vertical walls of a non-flat topography.
wall_adjustment = .T. limits the mixing length to a maximum of 1.8 *
z. This condition typically affects only the first grid level above the bottom boundary or near walls.In case of walls (non-flat topography), the respective horizontal distance from vertical walls is used instead of
z.
wall_heatflux#
- Fortran Type: R (6)
- Default: 0.0
-
SI-Unit: K m s-1
Prescribed kinematic sensible heat flux at the six topography faces.
The flux at the respective faces can be assigned as follows:
wall_heatflux index face orientation wall_heatflux(0) upward facing wall_heatflux(1) westward facing wall_heatflux(2) eastward facing wall_heatflux(3) southward facing wall_heatflux(4) northward facing wall_heatflux(5) downward facing This parameter applies only in case of a non-flat topography. Prescribing wall_heatflux additionally requires setting of surface_heatflux. Furthermore, please note that wall_heatflux(0) only describes the kinematic flux for upward facing surfaces at non-zero heights, because only those surfaces are assumed to represent walls. At zero-height (bottom surface of the model) the kinematic flux is given by surface_heatflux instead.
The parameter random_heatflux can be used to impose random perturbations on the internal surface heat flux array
surf%shfthat holds values given by surface_heatflux (at the bottom surface of the model) and wall_heatflux(0) (at the topography/wall top face).wall_heatflux is allowed to be set as runtime parameter as well. In case of a restart run, it then overwrites the setting from the previous run and the new values will be used for further restarts, as long as no new setting via the runtime parameter namelist is made.
wall_humidityflux#
- Fortran Type: R (6)
- Default: 0.0
-
SI-Unit: m s-1
Prescribed kinematic humidity flux at the six topography faces.
The flux at the respective faces can be assigned as follows:
wall_humidityflux index face orientation wall_humidityflux(0) upward facing wall_humidityflux(1) westward facing wall_humidityflux(2) eastward facing wall_humidityflux(3) southward facing wall_humidityflux(4) northward facing wall_humidityflux(5) downward facing This parameter applies only in case of a non-flat topography and humidity = .T.. Prescribing wall_humidityflux additionally requires setting of surface_waterflux. Furthermore, please note that wall_humidityflux(0) only describes the kinematic flux for upward facing surfaces at non-zero height, because only those surfaces are assumed to represent walls. At zero-height (bottom surface of the model) the kinematic flux is given by surface_waterflux instead.
wall_scalarflux#
- Fortran Type: R (6)
- Default: 0.0
-
SI-Unit: kg m-2 s-1 (particle flux) or ppm m s-1 (gaseous flux)
Prescribed scalar flux at the six topography faces.
The flux at the respective faces can be assigned as follows:
wall_scalarflux index face orientation wall_scalarflux(0) upward facing wall_scalarflux(1) westward facing wall_scalarflux(2) eastward facing wall_scalarflux(3) southward facing wall_scalarflux(4) northward facing wall_scalarflux(5) downward facing This parameter applies only in case of a non-flat topography and passive_scalar = .T.. Prescribing wall_scalarflux additionally requires setting of surface_scalarflux. Furthermore, please note that wall_scalarflux(0) only describes the kinematic flux for upward facing surfaces at non-zero height, because only those surfaces are assumed to represent walls. At zero-height (bottom surface of the model) the kinematic flux is given by surface_scalarflux instead.
y_shift#
- Fortran Type: I
-
Default: 0
Shift of boundary values at the inflow in y-direction to prevent occurence of persistent streaklike structures oriented along x-direction.
This feature alleviates the occurrence of persistent streaklike structures that can appear during simulations with neutral stratification and insufficient long model domain in streamwise direction when using lateral cyclic boundary conditions, or an insufficient long recycling width when using non-cyclic boundary conditions together with the turbulence recycling method. For a description of these structures, see Munters (2016). This method is only implemented for a mean flow in positive x-direction.
To prevent these structures in case of lateral cyclic boundary conditions (bc_lr = 'cyclic', bc_ns = 'cyclic') psolver = 'multigrid' is required.
The method shifts the values of all prognostic quantities at the left boundary by multiples (number of multiples given by y_shift) of a subdomain size along y. The shift is such that a point (at coordinate
y = yr) on the right domain boundary no longer corresponds with the point with the same y-coordinate on the left boundary, but instead with the point with the coordinate y = yr + y_shift * (ny + 1 ) / npey. Conversely, a point on the left boundary with coordinatey = ylcorresponds with the point on the right boundary with the coordinate y = yl - y_shift * (ny+1) / npey. Negative values and also those that exceed npey are allowed. They are mapped to the range [0,npey-1] internally.The following figure shows a scetch for npex = npey = 5 and y_shift = 1:
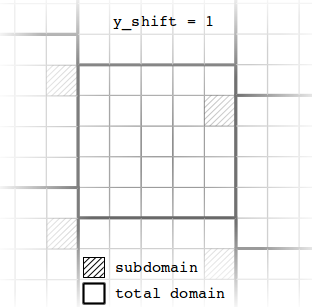
In case of using non-cyclic boundary conditions along x-direction (bc_lr = 'dirichlet/radiation') together with the turbulence recycling method (turbulent_inflow_method = 'recycle_...') the principal method is the same as explained above, except that the right boundary corresponds here to the position of the turbulence recycling plane.
zeta_max#
- Fortran Type: R
-
Default: 20.0
Upper limit of the flux Richardson Number.
Upper limit of the stability parameter \(\zeta= z_{mo} / L\) with \(z_{mo}\) the height of the constant flux layer and \(L\) the Obukhov length.
With a constant flux layer switched on (see constant_flux_layer), the Obukhov length \(L\) is calculated for \(z = z_{mo}\) in the 3d-model (in the 1d-model for all heights) for each horizontal grid point. Its particular values determine the values of the friction velocity (1d- and 3d-model) and the values of the eddy diffusivity (1d-model). With small wind velocities at the top of the constant flux layer or small vertical wind shears in the 1d-model, the stability parameter \(\zeta\) can take up unrealistic values. They are limited by an upper (zeta_max) and lower limit (see zeta_min). The condition zeta_max > zeta_min must be met.
zeta_min#
- Fortran Type: R
-
Default: -20.0
Lower limit of the flux Richardson Number.
Lower limit of the stability parameter \(\zeta= z_{mo} / L\) with \(z_{mo}\) the height of the constant flux layer and \(L\) the Obukhov length.
For further explanations see zeta_max. The condition zeta_max > zeta_min must be met.
z0h_factor#
- Fortran Type: R
-
Default: 1.0
Factor for calculating the roughness length for scalars.
With this parameter the roughness length for scalars (potential temperature, humidity/scalar) can be defined with respect to that for momentum. The roughness length for scalars z0h is calculated as:
z0h = z0h_factor * roughness_length.
This parameter is effective only in case that a constant flux layer is switched on at the bottom boundary (see constant_flux_layer).
Runtime Parameters#
averaging_interval#
- Fortran Type: R
- Default: undefined
-
SI-Unit: s
Averaging interval for all output of temporally averaged data.
This parameter defines the time interval length for temporally averaged data (vertical profiles, spectra, 2d cross-sections, 3d volume data). A value must be specified if output of averaged data is selected. The maximum interval length is limited by the value of parameter dt_data_output_av. In any case, averaging_interval <= dt_data_output_av must hold.
If an interval is defined, then by default the average is calculated from the data values of all time steps lying within this interval. The end point of the interval is always located at the output time as given by dt_data_output_av. The number of time levels entering into the average can be reduced via parameter dt_averaging_input.
If an averaging interval can not be completed at the end of a run, it will be finished at the beginning of the next restart run. Thus for restart runs, averaging intervals do not necessarily begin at the beginning of the run.
Parameters averaging_interval_pr and averaging_interval_sp can be used to define different averaging intervals for vertical profile data and spectra, respectively.
averaging_interval_pr#
- Fortran Type: R
- Default: Value of averaging_interval
-
SI-Unit: s
Averaging interval for output of vertical profiles to local file DATA_1D_PR_NETCDF.
If this parameter is given a non-zero value, temporally averaged vertical profile data are output. By default, profile data are not subject to temporal averaging. The maximum interval length is limited by the value of the parameter dt_dopr. In any case averaging_interval_pr <= dt_dopr must hold.
If an interval is defined, then by default the average is calculated from the data values of all time steps lying within this interval. The end point of the interval is always located at the output time as given by dt_dopr. The number of time levels entering into the average can be reduced via parameter dt_averaging_input_pr.
If an averaging interval can not be completed at the end of a run, it will be finished at the beginning of the next restart run. Thus for restart runs, averaging intervals do not necessarily begin at the beginning of the run.
bc_pt_b#
- Fortran Type: C*20
-
Default: dirichlet
Bottom boundary condition of the potential temperature at physical surfaces.
See initialization parameter bc_pt_b for further explanations.
cpu_log_barrierwait#
- Fortran Type: L
-
Default: .FALSE.
Sets an MPI-barrier at the beginning of each CPU time measurement.
Measurement of code performance is carried out by default for most parts of the PALM code (see routine
cpu_logfor more information). In case that MPI-calls are part of code segments to be measured, the measurement might be seriously affected by idle times (if MPI-calls on some of the cores have to wait because of other, previous MPI-calls on other cores which are not yet finished). In order to avoid measuring these idle times, you can switch on an MPI-barrier at the beginning of each measurement by setting cpu_log_barrierwait = .TRUE.. You should keep in mind that these additional barriers may generally degrade the code performance, so they should be switched on only for getting precise CPU time measurements (and not for production runs).
create_disturbances#
- Fortran Type: L
-
Default: .FALSE.
Imposes random perturbations to the horizontal velocity field.
With create_disturbances = .T., random perturbations can be imposed to the horizontal velocity field at certain times e.g. in order to trigger the onset of convection, etc..
The temporal interval between these times can be steered with dt_disturb, the vertical range of the perturbations with disturbance_level_b and disturbance_level_t, and the perturbation amplitude with disturbance_amplitude. In case of non-cyclic lateral boundary conditions (see bc_lr and bc_ns), the horizontal range of the perturbations is determined by inflow_disturbance_begin and inflow_disturbance_end. A perturbation is added to each grid point with its individual value determined by multiplying the disturbance amplitude with a uniformly distributed random number. As next steps, the arrays of
uandvare smoothed by applying a Shuman-filter twice, and then divergence is removed from the disturbed velocity field by applying the pressure solver.The random number generator to be used can be chosen via random_generator.
As soon as the desired flow features have developed (e.g. convection has started), further imposing of perturbations is not required and can be omitted (may not hold for non-cyclic lateral boundaries!). This can be steered by assigning an upper limit value for the perturbation energy (the perturbation energy is defined by the deviation of the velocity from the mean flow) using the parameter disturbance_energy_limit. As soon as the perturbation energy has exceeded this energy limit, random perturbations are added no more.
Timesteps where a random perturbation has been imposed are marked in the local file RUN_CONTROL by the character "D" which is appended to the values of the maximum horizontal velocities.
cross_profiles#
- Fortran Type: C*100 (100)
-
Default: [' u v ', ' theta ', ' w"theta" w*theta w*theta*BC wtheta wthetaBC ', ' w"u" w*u* wu w"v"w*v* wv ', ' km kh ', ' / ']*
Group variables in panels when plot script
palmplotis used.Determines which vertical profiles are to be presented in which coordinate system.
If output of vertical profiles is produced the appropriate data are written to a NetCDF file. Simultaneously, the model produces an attribute in the header of the NetCDF file which describes the layout for a plot to be generated with the plot software palmplot. The parameter cross_profiles determines how many coordinate systems (panels) the plot contains and which profiles are supposed to be drawn into which panel. (Currently, palmplot is limited to three profiles per panel.) cross_profiles expects a character string (up to 100 characters long) for each coordinate system, which consists of the names of the profiles to be drawn into this system (all available profiles and their respective names are described at parameter data_output_pr). The single names have to be separated by one blank (' ') and a blank must be spent also at the beginning and at the end of the string, e.g. cross_profiles = ' u v ', ' theta '.
In this case, the plot consists of two coordinate systems (panels) with the first panel containing the profiles of the horizontal velocity components of all output times (see dt_dopr) and the second one containing the profiles of the potential temperature.
Whether the coordinate systems are actually drawn, depends on whether data of the appropriate profiles were output during the run (profiles to be output have to be selected with the parameter data_output_pr). For example, if data_output_pr = 'u', 'v' was assigned, then the plot only consists of one panel, since no profiles of the potential temperature were output. On the other hand, if profiles were assigned to data_output_pr whose names do not appear in cross_profiles, this profiles will be plotted separately behind the profiles defined in cross_profiles.
The arrangement of the panels in the plot can be controlled via parameters profile_columns and profile_rows. Up to 100 panel systems are allowed in a plot (however, they may be put on several pages).
data_output#
- Fortran Type: C*30 (500)
-
Default: **
Quantities for which 2d cross section and/or 3d volume data are to be output.
Output of instantaneous data as well as of temporally averaged data is steered by strings assigned to this parameter (suffix
_avfor averaged quantities). See table of 3d output quantities for allowed values.Some of the quantities allow only 2d-output (horizontal sections). Their names must contain an asterisk
*at the end, e.g.pra*. See table of 2d output quantities for allowed values.All output files are in netCDF format. Information about the file content (kind of quantities, array dimensions and grid coordinates) are part of the self-describing netCDF format and can be extracted from the netCDF files using the command
ncdump -c <filename>. See netCDF data output for a list of the local filenames and their respective control parameters.Multiple quantities can be assigned, e.g. data_output = 'e', 'u', 'w' .
By assigning the pure strings of a 3d output quantity, volume data is output. Cross section data can be output by appending strings '_xy', '_xz', or '_yz' to the respective 3d quantity. Time-averaged output is generated by appending the string '_av' (for cross section data, this string must be appended after the cross section string). Cross section data can also be (additionally) averaged along the direction normal to the respective section (see below). Assignments of quantities can be given in arbitrary order:
Example:
data_output = 'u', 'theta_xz_av', 'w_xy', 'u_av'This example will create the following output: instantaneous 3d volume data of u-velocity component, temporally averaged 3d volume data of u-velocity component, instantaneous horizontal cross section data of w-velocity component, and temporally averaged vertical cross section data of potential temperature.
Output can be limited in the vertical direction up to a certain grid point via parameter nz_do3d.
Cross sections extend through the total model domain. In the two horizontal directions all grid points with
0 <= i <= nxand0 <= j <= nyare output. Be aware that in case of cyclic boundary conditions the respective opposite borders of the plots do not exactly match. The location(s) of the cross sections can be defined via parameters section_xy, section_xy_m, section_xz, section_xz_m, section_yz, and section_yz_m. Assigningsection_... =-1 causes the output data to be averaged along the direction normal to the respective section.Control of output times:
The time interval of the output times is determined via dt_data_output. This value is valid for all types of output quantities by default. Individual time intervals for instantaneous(!) 3d and section data can be declared using dt_do3d, dt_do2d_xy, dt_do2d_xz, and dt_do2d_yz.
Also, an individual time interval for output of temporally averaged data can be assigned using parameter dt_data_output_av. This applies to both 3d volume and cross section data. The length of the averaging interval is controlled via parameter averaging_interval. This parameter is obligatory if time averaged quantities have been selected.
The parameter skip_time_data_output can be used to shift data output activities for a given time interval. Individual intervals can be set using skip_time_do3d, skip_time_do2d_xy, skip_time_do2d_xz, skip_time_do2d_yz, and skip_time_data_output_av.
Output of user-defined quantities:
Besides the standard quantities from the above list, the user can output any other quantities. These have to be defined and calculated within the user-defined code (see User-defined output quantities). They can be selected for output with the user-parameter data_output_user for which the same rules apply as for data_output. Output of the user-defined quantities (time interval, averaging, selection of cross sections, etc.) is controlled by the parameters listed above and data are written to the same file(s) as the standard quantities.
Parallel output:
By default (netcdf_data_format = 2), for runs on more than one core (processor element, PE), the respective cores output only data of their respective subdomains into separate local files (file names are constructed by appending the five digit core ID, e.g.
<filename>_00000,<filename>_00001, etc.). After a run has been finished, the contents of these individual files are sampled into one final file using the helper routine combine_plot_fields.x, which is automatically called bypalmrun.Alternatively, PALM is able to collect all grid points of a cross section on PE0 before an output is done. In this case, only one output file (DATA_2D_XY_NETCDF, etc.) is created and combine_plot_fields.x is not invoked. In case of very large numbers of horizontal grid points, sufficient memory is required on PE0. This method can be activated via data_output_2d_on_each_pe = .F..
For netcdf_data_format < 5, 3d volume data output is handled separately by each core so that combine_plot_fields.x has to be called after the simulation has been finished. Be aware of extremely large execution times of combine_plot_fields.x, since it executes on one core only. Using netcdf_data_format > 4, output is written in parallel into one netCDF file during the run and combine_plot_fields.x is not invoked.
data_output_2d_on_each_pe#
- Fortran Type: L
-
Default: .TRUE.
Output 2d cross section data by one or all cores.
In runs with using several cores, by default, each core outputs cross section data of its subdomain into an individual file. After the simulation has finished, the contents of these files have to be sampled into one file using the helper routine combine_plot_fields.x.
Alternatively, via data_output_2d_on_each_pe = .F., the respective data is gathered on PE0 and output is done directly into one file, so combine_plot_fields.x does not have to be invoked. However, in case of very large numbers of horizontal grid points, sufficient memory is required on PE0.
data_output_masks#
- Fortran Type: C*30 (300,100)
-
Default: **
Quantities for which masked data are to be output.
Unlimited different masks can be defined (see masked data output). For each mask different instantaneous or temporally averaged quantities (up to 100) can be output. The masks are steered via parameters mask_x, mask_y, mask_z, mask_x_loop, mask_y_loop, mask_z_loop, and mask_k_over_surface. It is possible to scale the masked data with a specified length for each direction (see mask_scale_x, mask_scale_y, and mask_scale_z).
By default, masked data are output to local files
DATA_MASK_01_NETCDF, etc. If the user has switched on the output of temporally averaged data, these are written separately to local filesDATA_MASK_01_AV_NETCDF, etc. The file's format is netCDF. Further details about processing netCDF data are given in netCDF data output.See table of 3d mask output quantities for allowed quantities.
Example:
data_output_masks(1,:) = 'u', 'v', 'u_av', 'v_av', data_output_masks(2,:) = 'w', 'w_av', data_output_masks(3,:) = 'theta', 'u', 'v', 'w',This example will create the following output:
- Mask 1: Instantaneous data of u- and v-velocity component and temporally averaged data of u- and v-velocity component
- Mask 2: Instantaneous data of w-velocity component and temporally averaged data of w-velocity component
- Mask 3: Instantaneous data of potential temperature, u-, v- and w-velocity component
The user is allowed to extend the above list of quantities by defining his own output quantities (see the user-parameter data_output_masks_user).
The time intervals of the output times for each mask are determined via dt_domask. An individual time interval for output of temporally averaged data can be assigned using the parameter dt_data_output_av. The length of the averaging interval is controlled via parameter averaging_interval. This parameter is obligatory if time averaged quantities have been selected. No particular parameters are existent for steering the time-averaged output of each separate mask.
The parameter skip_time_domask can be used to shift data output activities for a given time interval.
By default, up to 300 different masks can be assigned (
max_masks = 300). If you wish to output more masks, change variablemax_masksin modules.f90 to the desired value. Ifmax_masksexceeds 999, format descriptors in check_open.f90 for filename creation need to be further adapted. This case, please contact the PALM developer team.
data_output_pr#
- Fortran Type: C*30 (500)
-
Default: **
Quantities for which vertical profiles (horizontally averaged) are to be output.
By default, vertical profile data is output to local netCDF file
DATA_1D_PR_NETCDF. Further details about processing of netCDF data are given in chapter netCDF data output.For horizontally averaged vertical profiles, always all vertical grid points (
0 <= k <= nz+1) are output to file. Vertical profile data refers to the total domain by default. But profiles for subdomains can also be output (see statistic_regions).The temporal interval of the output times of profiles is assigned via dt_dopr.
Profiles can also be temporally averaged (see averaging_interval_pr and dt_averaging_input_pr).
The list of available output quantities depends on the modules used for the simulation. See table of profile quantities for the entire list of all allowed quantities.
The profile data is either defined on u-v-levels (variables marked in red) or on w-levels (green). According to this, the z-coordinates of the individual profiles vary. Beyond that, with a constant flux layer switched on (see constant_flux_layer) the lowest output level is
z = zu(1)instead ofz = zw(0)for profiles w"u", w"v", wu and wv. Turbulence quantities such as w*u* or u*2 are calculated from turbulent fluctuations that are defined as deviations from the instantaneous horizontal average.Beyond that, initial profiles (at t = 0) of some variables can additionally be output. To trigger the output of initial profiles, a leading '#' must be added to the quantities' names. Possible values are:
#u, #v, #theta, #km, #kh, #l, #thetal, #q, #qv, #s, #sa, #thetav, #t_soil, #m_soilA leading '#' will not suppress output at later timesteps, i.e., quantities must not be specified twice with and without a '#'.
The user is allowed to extend the above list of quantities by defining his own output quantities (see the user-parameter data_output_pr_user).
Additionally, the user can define further output quantities within the user interface.
Note: The Kolmogorov length scale \(\eta\) (output quantity 'eta') is defined by \((\frac{\nu^3}{\epsilon})^{1/4}\) with \(\nu\) being the kinematic, molecular viscosity and \(\epsilon\) the turbulent kinetic energy dissipation rate. However, as LES is not resolving the smallest scales of the turbulent velocity field, the direct determination of the dissipation rate and therefore of the Kolmogorov length scale is not possible in LES mode. Therefore, the output quantity 'eta' has a meaningful value only if the simulation is run in DNS mode.
debug_output#
- Fortran Type: L
-
Default: .FALSE.
Switch for output of debugging information during initialization phase and final actions of the simulation.
By adding the parameter debug_output = .T., pre-defined debug_message(s) will be printed into separate files (one per core, using Fortran unit 9) in the temporary folder of a run, named
DEBUG_<core-id>, where<core-id>is a six digit integer. Debug information may be helpful to narrow down the location of a model crash simply by setting this parameter instead of having to add WRITE statements and re-compile the code. Once the location is narrowed down, you may add further messages viaCALL debug_message (....). See the source code for stringdebug_messageto find out how it works.Note:
AddingCALL debug_message (....)in new places might require adding:USE control_parameters, & ONLY: debug_output, debug_stringin the declaration section of the source code file, if not already added.
Additionally, the flag debug_output_timestep enables debug output during timestepping. Keep in mind that this may create very large debug files.
Important:
Do not forget the-Boption in thepalmruncall. Otherwise, the temporary job folder, in which theDEBUG_00*files are located, will be deleted after finishing the job.
debug_output_timestep#
- Fortran Type: L
-
Default: .FALSE.
Switch for output of debugging information during timestepping.
For further details, see debug_output.
disturbance_amplitude#
- Fortran Type: R
- Default: 0.25
-
SI-Unit: m s-1
Maximum perturbation amplitude of the random perturbations.
Since the perturbation procedure includes two filter operations, the amplitude assigned by disturbance_amplitude is only an approximate value of the real magnitude of the perturbation.
See parameter create_disturbances for how random perturbations are imposed to the horizontal velocity field.
disturbance_energy_limit#
- Fortran Type: R
- Default: 0.01
-
SI-Unit: m-2 s-2
Upper limit value of the perturbation energy.
The perturbation energy (resolved-scale turbulence kinetic energy) E* is defined as one half of the total domain volume average of the sum of the squares of the deviations of the velocity components from the mean flow (horizontal average) times. As soon as the perturbation energy exceeds the assigned value, random perturbations to the fields of horizontal velocities are imposed no more. The value of disturbance_energy_limit usually must be determined by trial and error (it depends e.g. on the total number of grid points).
Parameter create_disturbances describes how to impose random perturbations to the horizontal velocity field.
disturbance_level_b#
- Fortran Type: R
- Default: Value of zu(3) or zu(nzt*2/3)
-
SI-Unit: m
Lower limit of the vertical range for random perturbations.
This parameter must hold the condition
zu(3)<= disturbance_level_b <=zu(nzt-2). Additionally, disturbance_level_b <= disturbance_level_t must also hold.In case of ocean runs the default value is disturbance_level_b =
zu(nzt* 2/3)(negative).Parameter create_disturbances describes how to impose random perturbations to the horizontal velocity field.
disturbance_level_t#
- Fortran Type: R
- Default: Value of zu(nzt/3) or zu(nzt-3)
-
SI-Unit: m
Lower limit of the vertical range for random perturbations.
This parameter must hold the condition disturbance_level_t <=
zu(nzt-2). Additionally, disturbance_level_b <= disturbance_level_t must also hold.In case of ocean runs the default value is disturbance_level_t =
zu(nzt-3)(negative).Parameter create_disturbances describes how to impose random perturbations to the horizontal velocity field.
do2d_at_begin#
- Fortran Type: L
-
Default: .FALSE.
Output of 2d cross section data at the beginning of a run.
The temporal intervals of output times of 2d cross section data (see data_output) are usually determined via parameters dt_do2d_xy, dt_do2d_xz and dt_do2d_yz. By assigning do2d_at_begin = .T., an additional output will be made at the beginning of a run (thus at the time t = 0 or at the respective starting times of restart runs).
do3d_at_begin#
- Fortran Type: L
-
Default: .FALSE.
Output of 3d volume data at the beginning of a run.
The temporal intervals of output times of 3d volume data (see data_output) is usually determined via parameter dt_do3d. By assigning do3d_at_begin = .T. an additional output will be made at the beginning of a run (thus at the time t = 0 or at the respective starting times of restart runs).
dt#
- Fortran Type: R
- Default: Depends on CFL criterion
-
SI-Unit: s
Time step for the 3d-model.
See dt in initialization_parameters namelist.
dt_averaging_input#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: s
The interval at which data enters into a temporal averaging period.
By default, data from each time step within the interval defined by averaging_interval enter into the calculation of the temporal average. By choosing dt_averaging_input > dt, the number of time levels entering the average can be minimized. This reduces the CPU time of a run but may worsen the quality of the averages statistics.
With a variable time step (see dt), the number of time levels entering the average can vary from one averaging interval to the next (for a more detailed explanation see averaging_interval). It is approximately given by the ratio of averaging_interval / MAX( dt_averaging_input, dt), which gives a more or less exact value, if a fixed time step is used and if this is an integral divisor of dt_averaging_input.
Example:
With averaging_interval = 100.0 and dt_averaging_input = 10.0, the time levels entering the average have a (minimum) distance of 10.0 s. Their distance may, of course, be larger if the current time step is larger than 10.0 s, so the average is calculated from the data of (maximum) 10 time levels.It is allowed to change dt_averaging_input during a job chain. In such a case, if the last averaging interval of the run previous to the change could not be completed (i.e. has to be finished in the current run), the data entering the averaging are not uniformly distributed over the averaging interval.
Parameter dt_averaging_input_pr can be used to define a different temporal interval for vertical profile data and spectra.
dt_averaging_input_pr#
- Fortran Type: R
- Default: Value of dt_averaging_input
-
SI-Unit: s
The interval at which vertical profiles and/or spectra data enter into a temporal averaging period.
By default, data from each time step within the interval defined by averaging_interval_pr, and averaging_interval_sp enter into the calculation of the temporal average. By choosing dt_averaging_input_pr > dt, the number of time levels entering the average can be minimized. This reduces the CPU time of a run but may worsen the quality of the averages statistics.
For more explanations see parameter dt_averaging_input.
dt_coupling#
- Fortran Type: R
- Default: 9999999.9
-
SI-Unit: s
Temporal interval for the data exchange in case of runs with atmosphere / ocean coupling.
This parameter specifies the temporal interval at which data are exchanged at the interface between the atmosphere and the ocean model. dt_coupling must be explicitly specified in the parameter files for both coupled models. If values given for the atmosphere and the ocean don't match, the atmosphere value will be used for the ocean model. In order to ensure synchronous coupling throughout the simulation, dt_coupling should be chosen larger than dt_max.
dt_data_output#
- Fortran Type: R
- Default: 9999999.9
-
SI-Unit: s
Temporal interval at which data shall be output.
This concerns 3d volume data (instantaneous or time averaged), cross sections (instantaneous or time averaged), vertical profiles and spectra. If data output is switched on (see data_output, data_output_pr, data_output_sp, and section_xy, this parameter can be used to assign the temporal interval at which these data shall be output. Output can be skipped at the beginning of a simulation using parameter skip_time_data_output, which has zero value by default. The reference time is the beginning of the simulation, i.e. output takes place at times t = skip_time_data_output + dt_data_output, skip_time_data_output + 2*dt_data_output, skip_time_data_output + 3*dt_data_output, etc. Since output is only done at the discrete time levels given by the time step used, the actual output times can slightly deviate from these theoretical values.
Individual temporal intervals for the different output quantities can be assigned using parameters dt_do3d, dt_do2d_xy, dt_do2d_xz, dt_do2d_yz, dt_domask, dt_dopr, dt_dosp, and dt_data_output_av.
Warning:
In case of parallel netCDF I/O (netcdf_data_format >= 5), setting small values for dt_data_output may result in HDF errors during model initialization, since internal netCDF thresholds may be exceeded. This especially can happen if large 3d arrays are output.
dt_data_output_av#
- Fortran Type: R
- Default: Value of dt_data_output
-
SI-Unit: s
Temporal interval at which time averaged data shall be output.
If data output of time averaged 2d and 3d data is switched on (see data_output), this parameter can be used to assign the temporal interval at which they shall be output (see dt_dopr for calculation of actual output times). Output can be skipped at the beginning of a simulation using parameter skip_time_data_output_av.
The length of the averaging interval is controlled via parameter averaging_interval.
dt_disturb#
- Fortran Type: R
- Default: 9999999.9
-
SI-Unit: s
Temporal interval at which random perturbations are to be imposed on the horizontal velocity field.
See parameter create_disturbances for how random perturbations are imposed to the horizontal velocity field.
dt_domask#
- Fortran Type: R (300)
- Default: Value of dt_data_output
-
SI-Unit: s
Temporal interval at which instantaneous masked data shall be output.
If output of masked data is switched on (see data_output_masks), this parameter can be used to assign the temporal interval at which these data shall be output (see dt_dopr for calculation of actual output times). For each mask, a separate output time can be assigned.
Example:
dt_domask = 600.0, 1800.0, 300.0
means that output of mask 1 is done every 600s, of mask 2 every 1800s, and of mask 3 every 300s.
dt_dopr#
- Fortran Type: R
- Default: Value of dt_data_output
-
SI-Unit: s
Temporal interval at which data of vertical profiles shall be output.
If output of horizontally averaged vertical profiles is switched on (see data_output_pr), this parameter can be used to assign the temporal interval at which profile data shall be output. Output can be skipped at the beginning of a simulation using parameter skip_time_dopr. The reference time is the beginning of the simulation, thus t = 0, i.e. output takes place at times t = skip_time_dopr + dt_dopr, skip_time_dopr + 2*dt_dopr, skip_time_dopr + 3*dt_dopr, etc. Since profiles can not be calculated for times lying within a time step interval, the output times can deviate from these theoretical values. If a time step ranges from t = 1799.8 to t = 1800.2, then in the example above the output would take place at t = 1800.2. In general, the output always lie between t = 1800.0 and t = 1800.0 +
dt. If the model uses a variable time step (which is the default), these deviations from the theoretical output times will, of course, be different for each output time.In order to guarantee an output of profile data at the end of a simulation (see end_time) in any way, end_time should be equal or a little bit larger than the respective theoretical output time. For example, if dt_dopr = 900.0 and 3600.0 seconds are to be simulated, then end_time >= 3600.0 should be chosen.
Profiles to be output can be selected via parameter data_output_pr.
dt_dopr_listing#
- Fortran Type: R
- Default: 9999999.9
-
SI-Unit: s
Temporal interval at which data of vertical profiles shall be output in ASCII format.
This parameter can be used to assign the temporal interval at which profile data shall be output to local file LIST_PROFIL. See dt_dopr for further explanations.
Output data and format of file LIST_PROFIL are internally fixed. Profiles of the most important model variables are arranged in adjacent columns.
dt_dots#
- Fortran Type: R
- Default: Depends on dt_run_control, dt_dopr, dt_averaging_input_pr
-
SI-Unit: s
Temporal interval at which time series data shall be output.
If not explicitly given, dt_dots is defined via
IF ( averaging_interval_pr == 0.0 ) THEN dt_dots = MIN( dt_run_control, dt_dopr ) ELSE dt_dots = MIN( dt_run_control, dt_averaging_input_pr ) ENDIFThis limits the number of calculation of statistics and, hence, the computational costs.
The actual output intervals can deviate from dt_dots due to a varying timestep dt. To generate output at every timestep, set dt_dots = 0.0.
Time series data are output to the local netCDF file DATA_1D_TS_NETCDF.
Instead of allowing to choose the time series output quantities, time-series output always contains a fixed list of quantities which depends on the modules used in the simulation. The list of output quantities of the timeseries output is available here.
Additionally, the user can add own time-series quantities to the file by using the user interface.
By default, time-series data refer to the total domain. Data can also be output for user-defined subdomains (see statistic_regions). However, quantities
umax,vmax,wmax,div_old, anddiv_newalways present the values of the total model domain (even in the output of subdomains).
dt_do2d_xy#
- Fortran Type: R
- Default: Value of dt_data_output
-
SI-Unit: s
Temporal interval at which horizontal (xy) cross section data shall be output.
If output of horizontal cross sections is switched on (see data_output, section_xy, and section_xy_m), this parameter can be used to assign the temporal interval at which cross section data shall be output (see dt_dopr for calculation of actual output times). Output can be skipped at the beginning of a simulation using parameter skip_time_do2d_xy.
Parameter do2d_at_begin has to be used if an additional output is wanted at the start of a run (thus at time t = 0 or at the respective starting times of restart runs).
dt_do2d_xz#
- Fortran Type: R
- Default: Value of dt_data_output
-
SI-Unit: s
Temporal interval at which vertical (xz) cross section data shall be output.
If output of vertical (xz) cross sections is switched on (see data_output, section_xz, and section_xz_m), this parameter can be used to assign the temporal interval at which cross section data shall be output (see dt_dopr for calculation of actual output times). Output can be skipped at the beginning of a simulation using parameter skip_time_do2d_xz.
Parameter do2d_at_begin has to be used if an additional output is wanted at the start of a run (thus at time t = 0 or at the respective starting times of restart runs).
dt_do2d_yz#
- Fortran Type: R
- Default: Value of dt_data_output
-
SI-Unit: s
Temporal interval at which vertical (yz) cross section data shall be output.
If output of vertical (yz) cross sections is switched on (see data_output, section_yz, and section_yz_m), this parameter can be used to assign the temporal interval at which cross section data shall be output (see dt_dopr for calculation of actual output times). Output can be skipped at the beginning of a simulation using parameter skip_time_do2d_yz.
Parameter do2d_at_begin has to be used if an additional output is wanted at the start of a run (thus at time t = 0 or at the respective starting times of restart runs).
dt_do3d#
- Fortran Type: R
- Default: Value of dt_data_output
-
SI-Unit: s
Temporal interval at which 3d volume data shall be output.
If output of 3d-volume data is switched on (see data_output), this parameter can be used to assign the temporal interval at which 3d-data shall be output (see dt_dopr for calculation of actual output times). Output can be skipped at the beginning of a simulation using parameter skip_time_do3d.
Parameter do3d_at_begin has to be used if an additional output is wanted at the start of a run (thus at time t = 0 or at the respective starting times of restart runs).
Warning:
In case of parallel netCDF I/O (netcdf_data_format >= 5), setting small values for dt_do3d may result in HDF errors during model initialization, since internal netCDF thresholds may be exceeded. This especially can happen if large 3d arrays are output.
dt_max#
- Fortran Type: R
- Default: 20.0
-
SI-Unit: s
Maximum allowed value of the time step.
By default, the time step that is internally calculated from the different time step criteria is restricted to have a maximum value of 20 s. This might be fine for simulations of any kind of atmospheric flow but may have to be changed for other setups.
Attention:
Since the simulation is automatically aborted if the internally calculated timestep is less than 10-5 * dt_max, dt_max generally needs adjustment e.g. in case of very small grid spacings (for example in wind tunnel setups that use the spatial scales of the wind tunnel), because in such setups time steps are usually very small, too.
dt_restart#
- Fortran Type: R
- Default: undefined
-
SI-Unit: s
Temporal interval at which a new restart run is to be carried out.
For a description how to assign restart times manually see runtime parameter restart_time. dt_restart does not show any effect, if restart_time has not been set.
If a job chain is automatically finished (i.e. end_time is reached), and the run shall be continued via another chain by increasing end_time, the restart mechanism has to be activated again by setting (e.g.) restart_time = (end time of first chain) + dt_restart. Setting dt_restart only will have no effect.
For coupled atmosphere/ocean runs this parameter must be equal in both parameter files PARIN and PARIN_O.
dt_run_control#
- Fortran Type: R
- Default: 60.0
-
SI-Unit: s
Temporal interval at which run control output is to be made.
Run control information is output to the local ASCII-file RUN_CONTROL. At each output time, one line with information about the size of the time step, maximum speeds, total kinetic energy etc. is written to this file. The reference time is the beginning of the simulation, i.e. output takes place at times t = dt_run_control, 2*dt_run_control, 3*dt_run_control, etc., and always at the beginning of a model run (thus at the time t = 0 or at the respective starting times of restart runs). The actual output times can deviate from these theoretical values (see dt_dopr).
Output after each time step can be achieved via dt_run_control = 0.0.
dt_run_control_spinup#
- Fortran Type: R
- Default: 3600.0
-
SI-Unit: s
Temporal interval at which run control output is to be made during the spinup phase.
See spinup_time for more details about the spinup phase.
Run control information of the spinup phase is output to the local ASCII-file RUN_CONTROL. At each output time, one line with information about the size of the time step and the mean surface temperature (as calculated by the surface models) is written to this file. Output takes place at times t = dt_run_control_spinup, 2*dt_run_control_spinup, 3*dt_run_control_spinup, etc., and always at the beginning of the spinup phase.
Output after each time step of the spinup phase can be achieved via dt_run_control_spinup = 0.0.
interpolate_to_grid_center#
- Fortran Type: L
-
Default: .FALSE.
Flag for interpolation of 2D/3D output on grid center.
If interpolate_to_grid_center = .T., all 2D- and 3D-output of non-centered output variables (u, v, w, wu, wv, wv, wtheta, wq, ws) is interpolated onto the center of the Arakawa-C grid, where scalars are defined. The wall-neighbouring grid points are interpolated with zero. Due to the changed grid in x-, y- or z-direction, buildings appear to be smaller in the output than they are in the prognostic equations. Please note, in case of a precursor run where the yz output data shall be further processed with the preprocessor tool palm_cti, interpolate_to_grid_center = .T. is not allowed.
end_time#
- Fortran Type: R
- Default: 100.0
-
SI-Unit: s
End of simulated time.
The time to be simulated is starting from the beginning of the initialization run (t = 0), not starting from the beginning of the respective restart run.
For coupled atmosphere/ocean runs this parameter must be equal in both parameter files PARIN and PARIN_O.
force_print_header#
- Fortran Type: L
-
Default: .FALSE.
Steering of header output to the local file RUN_CONTROL.
By default, informations about the model parameters in use are output to the beginning of file RUN_CONTROL for initial runs only (these informations are identical to that which are output to the local file HEADER). With force_print_header = .T., these informations are also output to RUN_CONTROL for each restart run.
homogenize_surface_temperature#
- Fortran Type: L
-
Default: .FALSE.
Parameter to switch on the homogenization of surface temperature at the beginning of a run.
See initialization parameter homogenize_surface_temperature for further explanations.
mask_k_over_surface#
- Fortran Type: I (300,100)
-
Default: undefined
List of heights above terrain (as grid level index offset) for the terrain-following masked output.
Output is created for the given grid level index offset above terrain height. This creates a terrain-following output. mask_k_over_surface must be \(\geq\) 1.
A list of multiple grid level index offsets can be given. Each masked output can be given a different list.
If mask_k_over_surface is specified for a mask, mask_z and mask_z_loop are ignored for this mask. Also, mask_scale_z has no effect on values given by mask_k_over_surface.
Example:
mask_k_over_surface(1,:) = 2, 5,creates a terrain-following output for mask 1 and outputs values at the 2nd and 5th grid level above the terrain.
mask_scale_x#
- Fortran Type: R
-
Default: 1.0
Scaling factor for masked data output along x-direction.
Positions given in mask_x are scaled (multiplied) by mask_scale_x.
For scaling the masked data along y- and z-direction, use mask_scale_y and mask_scale_z, respectively.
Example:
Setting mask_scale_x = 10 and mask_x(1,:) = 50.0, 100.0, will result in masked output at x = 500.0 m and x = 1000.0 m.
mask_scale_y#
- Fortran Type: R
-
Default: 1.0
Scaling factor for masked data output along y-direction.
Positions given in mask_y are scaled (multiplied) by mask_scale_y.
For scaling the masked data along x- and z-direction, use mask_scale_x and mask_scale_z, respectively.
Example:
Setting mask_scale_y = 10 and mask_y(1,:) = 50.0, 100.0, will result in masked output at y = 500.0 m and y = 1000.0 m.
mask_scale_z#
- Fortran Type: R
-
Default: 1.0
Scaling factor for masked data output along z-direction.
Positions given in mask_z are scaled (multiplied) by mask_scale_z.
For scaling the masked data along x- and y-direction, use mask_scale_x and mask_scale_y, respectively.
Example:
Setting mask_scale_z = 10 and mask_z(1,:) = 50.0, 100.0, will result in masked output at z = 500.0 m and z = 1000.0 m.
mask_x#
- Fortran Type: R (300,100)
- Default: undefined
-
SI-Unit: m
List of x-coordinates of mask positions.
This parameter defines all positions along x-direction where quantities for masked data are to be output (see data_output_mask). For each mask, a separate list must be assigned.
Per default, data is output at every grid point along x-direction.
The given coordinates are scaled using mask_scale_x.
Instead of assigning individual positions, mask_x_loop allows to define multiple positions equally spaced along x-direction.
For output positions along y- and z-direction, see mask_y and mask_z, respectively.
Example:
mask_x(1,:) = 50.0, 100.0, 500.0, 550.0, 600.0 mask_x(2,:) = 1000.0This example will create outputs at x = 50 m and x = 100 m, etc. for mask 1 and at x = 1000 m for mask 2.
See the masked data-output documentation for further examples.
mask_x_loop#
- Fortran Type: R (300,3)
- Default: undefined
-
SI-Unit: m
Loop begin, end, and stride for x-coordinates of mask locations.
This parameter allows to define multiple equally-spaced output positions along x-direction for masked data output.
mask_x_loop contains of 3 values:
- first output position,
- last output position,
- distance between output positions.
If the given last output position cannot be reached from the first output position by adding multiples of the distance, the last calculated position smaller than the given last output position is the actual last output position.
Per default, every grid point along x-direction is used in the masked output.
For each mask a separate mask_x_loop must be assigned.
If mask_x is specified, mask_x_loop will be ignored.
Example:
mask_x_loop(1,:) = 0.0, 2010.0, 100.0,This example creates output positions every 100 m starting at x = 0 m until x = 2000 m for mask 1.
See the masked data-output documentation for further examples.
mask_y#
- Fortran Type: R (300,100)
- Default: undefined
-
SI-Unit: m
List of y-coordinates of mask positions.
This parameter defines all positions along y-direction where quantities for masked data are to be output (see data_output_mask). For each mask, a separate list must be assigned.
Per default, data is output at every grid point along y-direction.
The given coordinates are scaled using mask_scale_y.
Instead of assigning individual positions, mask_y_loop allows to define multiple positions equally spaced along y-direction.
For output positions along x- and z-direction, see mask_x and mask_z, respectively.
Example:
mask_y(1,:) = 50.0, 100.0, mask_y(2,:) = 1000.0,This example will create outputs at y = 50 m and y = 100 m for mask 1 and at y = 1000 m for mask 2.
See the masked data-output documentation for further examples.
mask_y_loop#
- Fortran Type: R (300,3)
- Default: undefined
-
SI-Unit: m
Loop begin, end, and stride for y-coordinates of mask locations.
This parameter allows to define multiple equally-spaced output positions along y-direction for masked data output.
mask_y_loop contains of 3 values:
- first output position,
- last output position,
- distance between output positions.
If the given last output position cannot be reached from the first output position by adding multiples of the distance, the last calculated position smaller than the given last output position is the actual last output position.
Per default, every grid point along y-direction is used in the masked output.
For each mask a separate mask_y_loop must be assigned.
If mask_y is specified, mask_y_loop will be ignored.
Example:
mask_y_loop(1,:) = 0.0, 2010.0, 100.0,This example creates output positions every 100 m starting at y = 0 m until y = 2000 m for mask 1.
See the masked data-output documentation for further examples.
mask_z#
- Fortran Type: R (300,100)
- Default: undefined
-
SI-Unit: m
List of z-coordinates of mask positions.
This parameter defines all positions along z-direction where quantities for masked data are to be output (see data_output_mask). For each mask, a separate list must be assigned.
Per default, data is output at every grid point along z-direction.
The given coordinates are scaled using mask_scale_z.
Instead of assigning individual positions, mask_z_loop allows to define multiple positions equally spaced along z-direction.
For output positions along x- and y-direction, see mask_x and mask_y, respectively.
Example:
mask_z(1,:) = 50.0, 100.0, 500.0, 550.0, 600.0 mask_z(2,:) = 1000.0This example will create outputs at the specified points for masks 1 and 2. Given height levels refer to the zu-grid. Height levels of quantities defined on the zw-grid are shifted by half a grid spacing (e.g. z = 75.0, 125.0, 525.0, 575.0, 625.0, if
dz= 50.0)).See the masked data-output documentation for further examples.
mask_z_loop#
- Fortran Type: R (300,3)
- Default: undefined
-
SI-Unit: m
Loop begin, end, and stride for z-coordinates of mask locations.
This parameter allows to define multiple equally-spaced output positions along z-direction for masked data output.
mask_z_loop contains of 3 values:
- first output position,
- last output position,
- distance between output positions.
If the given last output position cannot be reached from the first output position by adding multiples of the distance, the last calculated position smaller than the given last output position is the actual last output position.
Per default, every grid point along z-direction is used in the masked output.
For each mask a separate mask_z_loop must be assigned.
If mask_z is specified, mask_z_loop will be ignored.
Example:
mask_z_loop(1,:) = 0.0, 2010.0, 100.0,This example creates output positions every 100 m starting at z = 0 m until z = 2000 m for mask 1.
See the masked data-output documentation for further examples.
netcdf_data_format#
- Fortran Type: I
-
Default: 2
Data format of netCDF output files.
Important:
Setting netcdf_data_format > 2 requires a netCDF4 library and the preprocessor switch-D__netcdf4must be set (see options%cpp_options,%compiler_options, and%linker_optionsin the configuration file).Files with netCDF4 format cannot be read with netCDF3 libraries.
Parallel file support (netcdf_data_format > 4) additionally requires to set the preprocessor switch
-D__netcdf4_parallel.Warning:
In case of parallel I/O (netcdf_data_format \(\geq\) 5), setting small values for dt_data_output or dt_do3d may result in HDF errors during model initialization, since internal netCDF thresholds may be execeeded. This is likely to happen if large 3d arrays are output.Currently 6 choices are available:
-
1
netCDF classic format (filesize is limited to 2GB).
-
2
netCDF 64-bit-offset format (large file support, but single variables are still limited to 2GB).
-
3
netCDF-4 (HDF5) format (files can be as large as thr file system supports; unlimited variable size), without parallel I/O support.
-
4
netCDF-4 format, but with
NF90_CLASSIC_MODELbit set (some new features of netCDF4 are not available), without parallel I/O support. -
5
same as 3, but with parallel I/O support.
-
6
same as 4, but with parallel I/O support.
-
netcdf_deflate#
- Fortran Type: I
-
Default: 0
Data compression level for netCDF4/HDF5 output files.
The data compression must be given in the range 0 - 9, where 0 means no compression, and 9 highest compression. Typically, compression level 1 should be sufficient. Higher data compression rates require additional time. The compression only works for the netCDF4/HDF5 format in non-parallel mode (see netcdf_data_format).
normalizing_region#
- Fortran Type: I
-
Default: 0
Determines the statistic region from which the normalization quantities are calculated.
If output data of the horizontally averaged vertical profiles (see data_output_pr) is to be normalized, the respective normalization quantities are by default calculated from the averaged data of the total model domain (normalizing_region = 0) and are thus representative for the total domain. Instead of that, normalization quantities can also be calculated for a statistic region by assigning the respective statistic region ID to normalizing_region (see also statistic_regions). These quantities are then used for normalizing of all profiles (even for that of the total domain).
\(0 \leq\) normalizing_region \(\leq 9\) is required.
npex#
- Fortran Type: I
-
Default: Depends on nx, ny, nz, and total number of cores of a run
Number of cores along x-direction of the virtual PE grid.
For parallel runs, the total number of cores (processor elements, PEs) to be used is given by the palmrun-option
-X. By default, the model tries to generate a 2d PE grid (domain decomposition along x and y), of more or less square-shape. If, for example, 16 cores are assigned (-X 16), a 4 \(\times\) 4 PE grid is generated (npex = npey = 4). This choice is optimal for square total domains (nx = ny) because it minimizes the number of ghost points at the lateral boundaries of the subdomains. If nx and ny differ extremely, the PE grid should be manually adjusted using adequate values for npex and npey.Important:
The value of npex \(\times\) npey must exactly match the value assigned by the palmrun-option-X. Otherwise the run aborts with a corresponding error message.Explicit setting of npex is not allowed for psolver = 'poisfft_sm'.
npey#
- Fortran Type: I
-
Default: Depends on nx, ny, nz, and total number of cores of a run
Number of cores along y-direction of the virtual PE grid.
See npex for further explanations.
Explicit setting of npey is not allowed for psolver = 'poisfft_sm'.
nz_do3d#
- Fortran Type: I
-
Default: Value of nz + 1
Limits the output of 3d volume data along the vertical direction (grid level index k).
By default, data for all grid points along z are output. Using nz_do3d limits the output up to the given vertical grid level index. This can be used to reduce the amount of output data. It affects all output of 3d volume data specified via data_output.
open_debug_files#
- Fortran Type: L
-
Default: .TRUE.
Flag for opening debug files.
By default, debug files with local filenames
DEBUG_<PE-id>(internal Fortran unit 9, PE-id` is a six digit integer) will be opened for each core in the temporary working directory at the beginning of a run. In order to avoid the opening of very large number of files in case of runs on many cores, set open_debug_files = .F.. However, debug files are opened in any case, if debug_output has been switched on.
profile_columns#
- Fortran Type: I
-
Default: 3
Number of coordinate systems to be plotted in one row by plot software
palmplot(see also postprocessing with ncl).It determines the layout of plots of horizontally averaged profiles (data_output_pr) when plotted with the plot software
palmplot. Generally, the number and sequence of coordinate systems (panels) to be plotted on one page are determined by cross_profiles. profile_columns determines how many panels are to be arranged next to each other in one row (number of columns). The respective number of rows on a page is assigned by profile_rows. According to their order given by data_output_pr, the panels are arranged beginning in the top row from left to right and then continued in the following row.
profile_rows#
- Fortran Type: I
-
Default: 3
Number of rows of coordinate systems to be plotted on one page by plot software
palmplot(see also postprocessing with ncl).It determines the layout of plots of horizontally averaged profiles. See profile_columns for further explanations.
pt_surface_heating_rate#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: K h-1
Linear rate of surface temperature increase.
See initialization parameter pt_surface_heating_rate for further explanations.
pt_surface_initial_change#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: K
Change in surface temperature to be applied at the beginning of a 3d run.
See initialization parameter pt_surface_initial_change for further explanations.
restart_data_format#
- Fortran Type: C*20
-
Default: mpi_shared_memory
Binary format of the input and output restart files.
For details see decription of restart_data_format in the initialization parameters namelist.
restart_data_format_input#
- Fortran Type: C*20
-
Default: Value of restart_data_format
Binary format of the input restart file.
See restart_data_format for allowed values.
This parameter can also be used in the initialization parameters namelist.
restart_data_format_output#
- Fortran Type: C*20
-
Default: Value of restart_data_format
Binary format of the output restart file.
See restart_data_format for allowed values.
This parameter can also be used in the initialization parameters namelist.
restart_time#
- Fortran Type: R
- Default: undefined
-
SI-Unit: s
Simulated time after which a restart run is to be carried out.
The simulated time refers to the beginning of the initial run (t = 0), not to the beginning of the respective restart run. Restart runs can additionally be forced to be carried out in regular intervals using the runtime parameter dt_restart.
Note:
A successful operation of this parameter requires to set option-a " ... restart ..."in the palmrun-call of the respective run (see also Initialization and restart runs).The choice of restart_time or dt_restart does not override the automatic start of restart runs in case that the job runs out of CPU time.
For nesting and coupled atmosphere/ocean runs, the parameter value must be the same in all respective namelist files. If this is not the case, the value given for the root/atmosphere model will be used.
section_xy#
- Fortran Type: I (100)
-
Default: undefined
Position(s) (grid level index k) of 2d horizontal cross section(s).
If output of horizontal cross sections is selected (see data_output), section_xy can be used to define the position(s) of the cross section(s). Up to 100 positions can be selected by assigning section_xy the corresponding vertical grid level index/indices
kof the requested cross section(s). The exact location (height level) of the cross section depends on the variable for which the output is requested:zu(k)for scalars and horizontal velocities,zw(k)for the vertical velocity. Information about the exact location of the cross section in m is contained in the netCDF output file. Parameter section_xy_m can be used instead or in addition to section_xy, to give locations in cartesian coordinates (m) instead of grid indices.Assigning section_xy = -1 creates the output of horizontal cross sections averaged along
z. In the netCDF output file these (averaged) cross sections are given the z-coordinate -1.0.Assignments to section_xy do not effect the output of horizontal cross sections of variables
us*andtheta*and the liquid water pathlwp*. For these quantities always only one cross section (forz = zu(1)) is output.
section_xy_m#
- Fortran Type: R (100)
-
Default: undefined
Position(s) of 2d horizontal cross section(s) in m.
See section_xy for a description of output of horizontal cross sections.
In the output file, sections given by section_xy_m are appended to those given by section_xy.
section_xz#
- Fortran Type: I (100)
-
Default: undefined
Position(s) (grid index j) of 2d (xz) vertical cross section(s).
If output of
xzcross sections is selected (see data_output), this parameter can be used to define the position(s) of the cross section(s). Up to 100 positions of cross sections can be selected by assigning section_xz the corresponding grid index/indicesjof the requested cross section(s). The exact location (in y-direction) of the cross section is given byj*dyor(j-0.5)*dy, depending on which grid the output quantity is defined. However, in the netCDF output file, no distinction is made between the quantities andj*dyis used for all positions. Parameter section_xz_m can be used instead or in addition to section_xz, to give locations in cartesian coordinates (m) instead of grid indices.Assigning section_xz = -1 creates the output of vertical cross sections averaged along
y. In the netCDF output file these (averaged) cross sections are given the y-coordinate -1.0.
section_xz_m#
- Fortran Type: R (100)
-
Default: undefined
Position(s) of 2d (xz) vertical cross section(s) in m.
See section_xz for a description of output of vertical cross sections.
In the output file, sections given by section_xz_m are appended to those given by section_xz.
Note:
For nested domains positions have to be given in the coordinate framework of the root domain.
section_yz#
- Fortran Type: I (100)
-
Default: undefined
Position(s) (grid index i) of 2d (yz) vertical cross section(s).
If output of
yzcross sections is selected (see data_output), this parameter can be used to define the position(s) of the cross section(s). Up to 100 positions of cross sections can be selected by assigning section_yz the corresponding grid index/indicesiof the requested cross section(s). The exact location (in x-direction) of the cross section is given byi*dxor(i-0.5)*dx, depending on which grid the output quantity is defined. However, in the netCDF output file, no distinction is made between the quantities andi*dxis used for all positions. Parameter section_yz_m can be used instead or in addition to section_yz, to give locations in cartesian coordinates (m) instead of grid indices.Assigning section_yz = -1 creates the output of vertical cross sections averaged along
x. In the netCDF output file these (averaged) cross sections are given the x-coordinate -1.0.
section_yz_m#
- Fortran Type: R (100)
-
Default: undefined
Position(s) of 2d (yz) vertical cross section(s) in m.
See section_yz for a description of output of vertical cross sections.
In the output file, sections given by section_yz_m are appended to those given by section_yz.
Note:
For nested domains positions have to be given in the coordinate framework of the root domain.
skip_time_data_output#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: s
No data output before this interval has passed.
Data output activities are starting not before the simulated time has reached the value given by skip_time_data_output (counting from the beginning of the simulation, t = 0). By default, this applies for output of instantaneous 3d volume data, cross section data, spectra and vertical profile data as well as for temporally averaged 2d and 3d data. Individual times can be assigned using parameters skip_time_do3d, skip_time_do2d_xy, skip_time_do2d_xz, skip_time_do2d_yz, skip_time_dosp, skip_time_dopr and skip_time_data_output_av.
Example:
For dt_data_output = 3600.0 and skip_time_data_output = 1800.0, the first output will be done at t = 5400 s.
skip_time_data_output_av#
- Fortran Type: R
- Default: Value of skip_time_data_output
-
SI-Unit: s
No output of temporally averaged 2d/3d data before this interval has passed.
skip_time_domask#
- Fortran Type: R (300)
- Default: Value of skip_time_data_output
-
SI-Unit: s
No output of masked data before this interval has passed.
skip_time_dopr#
- Fortran Type: R
- Default: Value of skip_time_data_output
-
SI-Unit: s
No output of vertical profile data before this interval has passed.
skip_time_do2d_xy#
- Fortran Type: R
- Default: Value of skip_time_data_output
-
SI-Unit: s
No output of instantaneous horizontal cross section data before this interval has passed.
skip_time_do2d_xz#
- Fortran Type: R
- Default: Value of skip_time_data_output
-
SI-Unit: s
No output of instantaneous vertical (xz) cross section data before this interval has passed.
skip_time_do2d_yz#
- Fortran Type: R
- Default: Value of skip_time_data_output
-
SI-Unit: s
No output of instantaneous vertical (yz) cross section data before this interval has passed.
skip_time_do3d#
- Fortran Type: R
- Default: Value of skip_time_data_output
-
SI-Unit: s
No output of instantaneous 3d volume data before this interval has passed.
surface_heatflux#
- Fortran Type: R
- Default: undefined
-
SI-Unit: depends on 'flux_input_mode'
Sensible heat flux at the bottom surface.
See initialization parameter surface_heatflux for further explanations.
synchronous_exchange#
- Fortran Type: L
-
Default: .FALSE.
Defines how MPI handles the exchange of ghost points.
By default, asynchronous transfer via
MPI_ISENDandMPI_IRECVis used. In case of synchronous_exchange = .T.,MPI_SENDRECVis used instead. On most networks the asynchronous method will give better performance.
termination_time_needed#
- Fortran Type: R
-
Default: 35.0
CPU time needed for terminal actions at the end of a run in batch mode.
If the string "restart" is given via
palmrunoption-a(see Initialization and restart runs), the remaining CPU time of the job is checked after each time step. Time stepping is stopped if the remaining CPU time is less than the value given by termination_time_needed.The integration of the prognostic equations must not consume the CPU time completely, since several actions still have to be carried out after the integration has been finished (e.g. writing of binary data for the restart run, carrying out output commands, copying of local files to their permanent destinations, etc.), which also takes some time. Furthermore, the parameter has also to account for the CPU time consumed by the job before the model has started (e.g. required for providing input data, or for compiling the user-interface code, etc.). The maximum possible time needed for these activities plus a reserve has to be given with the parameter termination_time_needed. Among other things, this time depends on the number of grid points used. If its value is selected too small, then the respective job will be prematurely aborted by the batch system, which may result in a data loss, and which will possibly interrupt the job chain.
The model aborts anyway, if the string "restart" is not given via
palmrunoption-a, and if the job has been assigned an insufficient CPU time viapalmrunoption-t.
wall_heatflux#
- Fortran Type: R (6)
- Default: 0.0
-
SI-Unit: K m s-1
Prescribed kinematic sensible heat flux at the six topography faces.
See initialization parameter wall_heatflux for further explanations.
Agent Parameters#
a_rand_target#
- Fortran Type: L (100)
-
Default: .FALSE.
Flag for agents within a group to have random targets outside of model domain.
There is one logical value per agent group (see number_of_agent_groups. If .T., each agent in that group will have a random target just outside the model domain. Otherwise, a target (see at_x/at_y) must be set manually for each agent group.
adx#
- Fortran Type: R (100)
- Default: 9999999.9
-
SI-Unit: m
Distance along x between agents within an agent source.
Each agent source is defined as an area from x = asl to asr and y = ass to asn. The first agent is released at x = asl; y = ass. More agents are released at x = asl + N * adx (x <= asr) and y = ass + N * ady (y <= asn), forming a raster.
To add a random element to these release points, see random_start_position_agents.
ady#
- Fortran Type: R (100)
- Default: 9999999.9
-
SI-Unit: m
Distance along y between agents within an agent source.
For further explanations see adx.
agent_maximum_age#
- Fortran Type: R
- Default: 9999999.9
-
SI-Unit: s
Maximum time an agent can spend in the simulation before it is deleted.
agent_time_unlimited#
- Fortran Type: L
-
Default: .FALSE.
time dimension in netCDF output file
By default, the time dimension in the netCDF output file for agent data is calculated by
MIN( multi_agent_system_end, end_time ) - multi_agent_system_start ) / ( dt_write_agent_data * 1.1
which adds additional 10% time levels as a safety factor. For agent_time_unlimited = .T., the time dimension will be defined as unlimited, so the netCDF file will contain exactly as many time levels as required for output. This minimizes the filesize, but may slow down the I/O performance for writing agent data.
alloc_factor_mas#
- Fortran Type: R
-
Default: 20.0
Factor (in percent) describing the additional memory allocated to the memory needed for initial agents at a given grid cell.
Implementation is the same as for the particle alloc_factor.
asl#
- Fortran Type: R (100)
- Default: 9999999.9
-
SI-Unit: m
Left border of agent source(s).
Check adx for information on agent sources.
asn#
- Fortran Type: R (100)
- Default: 9999999.9
-
SI-Unit: m
North border of agent source(s).
Check adx for information on agent sources.
asr#
- Fortran Type: R (100)
- Default: 9999999.9
-
SI-Unit: m
Right border of agent source(s).
Check adx for information on agent sources.
ass#
- Fortran Type: R (100)
- Default: 9999999.9
-
SI-Unit: m
South border of agent source(s).
Check adx for information on agent sources.
at_x#
- Fortran Type: R (100)
- Default: undefined
-
SI-Unit: m
x-coordinate of agent group target.
Each agent in a group will navigate through building topography toward the corresponding target with coordinate (x,y) = (at_x, at_y).
at_y#
- Fortran Type: R (100)
- Default: undefined
-
SI-Unit: m
y-coordinate of agent group target.
Each agent in a group will navigate through building topography toward the corresponding target with coordinate (x,y) = (at_x, at_y).
bc_mas_lr#
- Fortran Type: C*15
-
Default: absorb
Boundary condition for agents at the left and right model boundary.
Currently 2 choices are available:
-
absorb
Agents are deleted when they leave the model domain.
-
cyclic
Agents leaving the domain will enter it again from the opposite side.
-
bc_mas_ns#
- Fortran Type: C*15
-
Default: absorb
Boundary condition for agents at the north and south model boundary.
Currently 2 choices are available:
-
absorb
Agents are deleted when they leave the model domain.
-
cyclic
Agents leaving the domain will enter it again from the opposite side.
-
coll_t_0#
- Fortran Type: R
- Default: 3.0
-
SI-Unit: s
Range of unscreened interactions for social force model.
Collision avoidance of pedestrians is implemented according to the approach of Karamouzas et al. (2014). This parameter implies a maximum range that pedestrians consider for collision avoidance, i.e., potential collisions that will occur more than coll_t_0 seconds in the future are largely ignored.
NOTE: This parameter should generally not be changed.
corner_gate_start#
- Fortran Type: R
- Default: 0.5
-
SI-Unit: m
Distance of navigation gate starting point to obstacle corner.
Convex obstacle corners are used for agents to navigate toward their target (see at_x/at_y). To avoid collisions with these obstacles, the navigation points do not coincide exactly with the obstacle corners but are shifted outward along the bisector of the corner. The navigation point for each agent at a given corner is chosen randomly along a line following the corner bisector starting at corner_gate_start outward from the corner and ending at corner_gate_start + corner_gate_width outward from the corner, forming a "gate" at each obstacle corner through which agents can pass. This gate helps to avoid clustering of agents moving in different directions at corner navigation points.
corner_gate_width#
- Fortran Type: R
- Default: 1.0
-
SI-Unit: m
Width of navigation gate at obstacle corners.
See corner_gate_start for details.
deallocate_memory_mas#
- Fortran Type: L
-
Default: .TRUE.
Parameter to enable/disable deallocation of unused memory.
If the number of agents in a grid box exceeds the allocated memory, new memory is allocated. However, if the number of agents per grid box is only temporarily high, most of the memory will be unused later. If deallocate_memory_mas = .T., the allocated memory used for agents will be dynamically adjusted with respect to the current number of agents every step_dealloc_mas'th timestep.
dim_size_agtnum_manual#
- Fortran Type: I
-
Default: undefined
Manually set size for agent-number dimension.
Agent output has two dimensions, time and agent number. By default, the size of the latter is estimated via the output interval and amount, the MAS simulation time and agent_maximum_age. This is a maximum estimation and may thus be much too large, i.e. it may lead to a huge output filesize. Use dim_size_agtnum_manual to explicitly set the dimension size.
If dim_size_factor_agtnum is also set, the minimum of the resulting dimension sizes is chosen.
WARNING: This may lead to the dimension being smaller than the number of agents and thus data loss.
dim_size_factor_agtnum#
- Fortran Type: R
-
Default: 1.0
Factor to modify the agent-number dimension size for agent output.
Agent output has two dimensions, time and agent number. By default, the size of the latter is estimated via the output interval and amount, the MAS simulation time and agent_maximum_age. This is a maximum estimation and may thus be much too large, i.e. it may lead to a huge output filesize. Use dim_size_factor_agtnum to reduce the dimension size.
If dim_size_agtnum_manual is also set, the minimum of the resulting dimension sizes is chosen.
WARNING: This may lead to the dimension being smaller than the number of agents and thus data loss.
dist_to_int_target#
- Fortran Type: R
- Default: 0.25
-
SI-Unit: m
Distance at which an intermittent target counts as reached.
Each agent in an agent group navigates toward the target specficied for that group (at_x/at_y). To do this, a preprocessed navigation mesh (visibility graph) is searched for via the A*-algorithm to find the most efficient path to the target. The agent successively navigates toward a number of intermittent targets along the calculated path, each such target providing the direction of the driving force until it is reached. Each intermittent target counts as reached as soon as the distance between it and the current agent position is smaller than dist_to_int_target. Then, the next intermittent target along the path is chosen.
NOTE: This parameter should be chosen in a way that insures that the next intermittent target is visible to the agent once the current one is reached. If not, agents could get stuck at obstacle corners.
dt_agent#
- Fortran Type: R
- Default: 0.02
-
SI-Unit: s
Agent timestep.
The MAS performs a number of subtimesteps usually much smaller than the model timestep until all agents have reached the model timestep. The value of this parameter should not exceed 0.02 s, because above that, the local interaction of the agents will become unstable.
dt_arel#
- Fortran Type: R
- Default: 9999999.9
-
SI-Unit: s
Interval at which agents are released at their respective sources.
Every dt_arel seconds, agents are released at their sources as defined by asl, asr, adx, ass, asn and ady.
dt_write_agent_data#
- Fortran Type: R
- Default: 9999999.9
-
SI-Unit: s
Output interval for agent NetCDF output.
Agent data is output every dt_write_agent_data seconds to the NetCDF file DATA_AGT_NETCD.
Note: To animate the results of an agent simulation at real-time speed, dt_write_agent_data = 0.04 is recommended.
Note: While prognostic quantities are output at every output step, this data will not change between two PALM timesteps, because no new values have been calculated yet.
end_time_arel#
- Fortran Type: R
- Default: undefined
-
SI-Unit: s
Simulation time at which the release of agents is stopped.
From this time on no more agents are released from the sources as defined by asl, asr, adx, ass, asn and ady. There may still be agents left in the simulation until the last agent has reached agent_maximum_age and is deleted or until all agents have left the model domain or reached their targets.
max_dist_from_path#
- Fortran Type: R
- Default: 0.25
-
SI-Unit: m
Agent distance from current path section that triggers recalculation of the path of that agent.
The path of an agent to the target it is assigned to consists of a number of intermittent targets. If an agent deviates farther from current path section it is on (the line between its last and its current intermittent target) than max_dist_from_path, a recalculation of the path to the target it is assigned to is triggered.
min_nr_agent#
- Fortran Type: I
-
Default: 2
Minimum number of agents for which memory is allocated at every grid cell.
multi_agent_system_end#
- Fortran Type: R
- Default: 9999999.9
-
SI-Unit: s
Simulation time at which the multi agent system will stop executing.
NOTE: This does not consider spin-up time.
multi_agent_system_start#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: s
Simulation time at which the multi agent system will start executing.
NOTE: This does not consider spin-up time.
number_of_agent_groups#
- Fortran Type: I
-
Default: 1
Number of agent groups.
Sources (asl, asr, adx, ass, asn and ady ) and targets (at_x/at_y) should be defined for each group individually. A maximum of 100 groups is allowed.
Note: dt_arel is the same for all groups.
radius_agent#
- Fortran Type: R
- Default: 0.25
-
SI-Unit: m
Quasi-solid core of agents.
If agents collide, (distance of agent centers are smaller than 2 * radius_agent) the standard social force model according to Helbing and Molnár (1995) is used to calculate the repulsive forces they exert on each other, instead of the default collision avoidance scheme.
random_start_position_agents#
- Fortran Type: L
-
Default: .TRUE.
Initial position of the particles is varied randomly within certain limits.
By default, the initial positions of agents within the source exactly correspond with the positions given by asl, asr, adx, ass, asn and ady. With random_start_position_agents = .T., the initial positions of the agents are allowed to randomly vary from these positions within certain limits.
Very important: If random_start_position_agents = .T., the random-number generators on the individual PEs no longer run synchronously. If random disturbances are applied to the velocity field (see create_disturbances, then as a consequence for parallel runs, the realizations of the turbulent flow fields will deviate between runs in which different numbers of PEs are used!
read_agents_from_restartfile#
- Fortran Type: L
-
Default: .FALSE.
Flag to read agent data from restart file.
This currently has no function, because restart runs with agents are not yet implemented. Will follow.
repuls_agent#
- Fortran Type: R
- Default: 1.5
-
SI-Unit: m2 s-2
Magnitude of the repulsive force agents exert on each other.
The repulsive force agents exert on each other according to the original social force model Helbing and Molnár (1995) is calculated from a potential field and drops exponentially with distance. This is the magnitude of that potential field.
repuls_wall#
- Fortran Type: R
- Default: 7.0
-
SI-Unit: m2 s-2
Magnitude of the repulsive force obstacles exert on agents.
The repulsive force obstacles exert on agents according to the original social force model Helbing and Molnár (1995) is calculated from a potential field and drops exponentially with distance. This is the magnitude of that potential field.
scan_radius_agent#
- Fortran Type: R
- Default: 3.0
-
SI-Unit: m
Radius around the agent in which it scans for obstacles.
Based on scan_radius_agent, each agent at each subtimestep determines a number of gridboxes around it and scans them for obstacles (topography and other agents). It will interact only with obstacles in that radius. The limiting factor is the grid spacing, because a maximum of three grid boxes can be scanned at subdomain boundaries.
sigma_rep_agent#
- Fortran Type: R
- Default: 0.3
-
SI-Unit: m
Width of agent repulsive field.
The repulsive force agents exert on each other according to the original social force model Helbing and Molnár (1995) is calculated from a potential field and drops exponentially with distance. This is the width of that potential field.
sigma_rep_wall#
- Fortran Type: R
- Default: 0.1
-
SI-Unit: m
Width of obstacle repulsive field.
The repulsive force obstacles exert on agents according to the original social force model Helbing and Molnár (1995) is calculated from a potential field and drops exponentially with distance. This is the width of that potential field.
step_dealloc_mas#
- Fortran Type: I
-
Default: 100
Interval for memory reallocation.
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Parameter to switch on/off the module in general.
switch_off_module = .T. can be used to switch off this module without removing (or commenting) the namelist from the namelist file.
tau_accel_agent#
- Fortran Type: R
- Default: 0.5
-
SI-Unit: s
Relaxation time in social force model.
tau_accel_agent determines how quickly an agent will approach its desired velocity and direction. A smaller value leads to a more aggresive walking style.
Biometeorology Parameters#
thermal_comfort#
- Fortran Type: L
-
Default: .FALSE.
Switches on/off the calculation of thermal comfort indices.
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Switches on/off the entire module.
If .T., the biometeorology module is not activated even if the namelist is present in the parameter file.
Bulk Cloud Parameters#
aerosol_bulk#
- Fortran Type: C*20
-
Default: nacl
Defines the type of aerosol to use.
The molecular weight, denisty and the solubility (vant Hoff factor) of each species is considered.
Currently 3 choices are available:
-
nacl
Sea salt is assumed as the bulk aerosol.
-
c3h4o4
Malonic acid is assumed as the bulk aerosol.
-
nh4no3
Ammonium sulfate is assumed as the bulk aerosol.
-
c_sedimentation#
- Fortran Type: R
-
Default: 2.0
Courant number for sedimentation process.
A Courant number that is too large inhibits microphysical interactions of the sedimented quantity. There is no need to set limiter_sedimentation if c_sedimentation \(\leq\) 1.0. This parameter only comes into effect if cloud_scheme = 'morrison' or 'seifert_beheng'.
call_microphysics_at_all_substeps#
- Fortran Type: L
-
Default: .FALSE.
Controls how frequently two-moment cloud microphysics are computed.
Using the default, cloud microphysics are computed once before the time step. When set to .T., cloud microphysics are computed before every substep of the applied time step scheme. This is, however, unnecessary to obtain acceptable results. Note that advection and diffusion of rainwater mixing ratio (qr) and rain drop concentration (nr) are not affected by this parameter (these processes are computed as any other scalar).
cloud_scheme#
- Fortran Type: C*20
-
Default: saturation_adjust
Defines cloud microphysics for bulk cloud physics.
Currently 5 choices are available:
-
kessler
One-moment cloud microphysics according to Kessler (1969). It is also uses the saturation adjustment scheme to diagnose cloud water. However, it allows precipitation if the liquid cloud water exceeds a threshold value. This water is instantaneously removed from the model domain. Additionally, liquid cloud water is allowed to sediment if cloud_water_sedimentation = .T..
-
morrison
Two-moment cloud microphysics according to Seifert and Beheng (2006), Khairoutdinov and Kogan (2000), Khvorostyanov and Curry (2006) and Morrison and Grabowski (2007). This scheme is an extension of the 'seifert_beheng' scheme. However, there are three main differences. First, instead of saturation adjustment, the diffusional growth is parametrized while calculating condensation/evaporation rates, explicitly. For appropriate usage of this scheme, the time step must be smaller than the diffusional growth relaxation time. Usually, this is on the order of 1-2 seconds. Second, the activation is considered with a simple Twomey activation-scheme. Koehler-theory accounts for this with the parameter curvature_solution_effects_bulk = .T.. The background aerosol concentration, which determines the maximum number of activated cloud droplets, can be prescribed with na_init. Thirdly, the number concentration of cloud droplets (
nc) and the cloud water mixing ratio (qc) are prognostic quantities. -
morrison_no_rain
Two-moment cloud microphysics according to Seifert and Beheng (2006), Khairoutdinov and Kogan (2000), Khvorostyanov and Curry (2006) and Morrison and Grabowski (2007). The 'morrison' scheme is applied as a stand-alone module, i.e. the rain processes of Seifert and Beheng are neglected and
qrandnrremain unallocated. -
saturation_adjust
Simple saturation adjustment scheme (also known as 0%-or-100% scheme) in which a grid volume is either saturated or subsaturated. Detailed information about the condensation scheme is given in the description of the cloud physics module. Supersaturations are instantaneously condensed to liquid water. No precipitation is produced. If precipitation is important, use 'kessler', 'morrison' or 'seifert_beheng'.
-
seifert_beheng
Two-moment cloud microphysics according to Seifert and Beheng (2006). It uses the 'saturation_adjustment' scheme to diagnose cloud water. The cloud drop number concentration is set via nc_const. Rain water, and hence precipitation, is treated with two additional prognostic equations for rain water mixing ratio and rain drop concentration that include autoconversion, accretion, selfcollection, breakup, evaporation, and sedimentation. The effect of ventilation on evaporation is steered by ventilation_effect. Sedimentation is controlled via c_sedimentation or limiter_sedimentation. The effects of turbulence on accretion and autoconversion are steered via collision_turbulence. Additionally, liquid cloud water is allowed to sediment if cloud_water_sedimentation = .T..
-
cloud_water_sedimentation#
- Fortran Type: L
-
Default: .FALSE.
Enables/disables sedimentation of cloud water according to Ackerman et al. (2009).
This parameter is only used when cloud_scheme = 'kessler', 'morrison' or 'seifert_beheng'.
collision_turbulence#
- Fortran Type: L
-
Default: .FALSE.
Enables/disables effects of turbulence on the collision process according to Seifert, Nuijens and Stevens (2010).
This parameter is only used when cloud_scheme = 'morrison' or 'seifert_beheng'.
curvature_solution_effects_bulk#
- Fortran Type: L
-
Default: .FALSE.
Enables/disables effects of curvature and solution on cloud droplet activation.
The parameterization of Khvorostyanov and Curry (2006) is used. The physio-chemical aerosol properties are prescribed with aerosol_bulk, dry_aerosol_radius and sigma_bulk.
dry_aerosol_radius#
- Fortran Type: R
- Default: 5e-08
-
SI-Unit: m
The mean geometric radius of the dry aerosol spectrum.
graupel#
- Fortran Type: L
-
Default: .FALSE.
Enables/disables calculation of prognostic equations for graupel.
If set to .T., will compute the graupel mixing ratio and number concentration. This is only allowed if snow = .T., if cloud_scheme = 'morrison' or 'seifert_beheng' and microphysics_ice_phase = .T..
ice_crystal_sedimentation#
- Fortran Type: L
-
Default: .FALSE.
Enables/disables sedimentation of ice crystals according to Seifert and Beheng (2006).
This parameter can only be set .T. if cloud_scheme = 'morrison' or 'seifert_beheng' and microphysics_ice_phase = .T..
in_init#
- Fortran Type: R
- Default: 1000.0
-
SI-Unit: m-3
Background ice nuclei concentration.
limiter_sedimentation#
- Fortran Type: L
-
Default: .TRUE.
Enables/disables slope limiter in sedimentation process according to Stevens and Seifert (2008).
This parameter is only used if cloud_scheme = 'morrison' or 'seifert_beheng'. If c_sedimentation \(\leq\) 1.0 there is no need to use the limiter.
microphysics_ice_phase#
- Fortran Type: L
-
Default: .FALSE.
Enables/disables calculation of prognostic equations for ice crystals.
If set to .T., will compute the ice crystal mixing ratio and number concentration. If temperatures allow, ice crystals can form via nucleation and grow due to water vapor deposition. Liquid phase is considered by default. Currently, mixed-phase microphysics is only supported for snow_scheme = 'morrison' and 'seifert_beheng'. Furthermore, graupel and snow require microphysics_ice_phase = .T. as a condition.
na_init#
- Fortran Type: R
- Default: 100000000.0
-
SI-Unit: m-3
Background dry aerosol concentration.
If cloud_scheme = 'morrison' is used, this parameter replaces nc_const. Activation is parameterized assuming that the number of activated cloud condensation nuculei (CCN) cannot be larger than na_init. This parameter is only used if cloud_scheme = 'morrison'.
nc_const#
- Fortran Type: R
- Default: 70000000.0
-
SI-Unit: m-3
Constant cloud droplet number density.
The default value is applicable for marine conditions. This parameter is only used if cloud_scheme = 'seifert_beheng'.
precipitation_amount_interval#
- Fortran Type: R
- Default: Value of dt_do2d_xy
-
SI-Unit: s
Temporal interval for which the precipitation amount (in mm) shall be calculated and output.
This parameter requires cloud_scheme = 'kessler' or 'seifert_beheng'. The interval must be smaller or equal than the output interval for 2d horizontal cross sections given by dt_do2d_xy. The output of the precipitation amount also requires setting of data_output = 'pra*'.
sigma_bulk#
- Fortran Type: R
-
Default: 2.0
The dispersion of the dry aerosol spectrum.
snow#
- Fortran Type: L
-
Default: .FALSE.
Enables/disables calculation of prognostic equations for snow.
If set to .T., will compute the snow mixing ratio and number concentration. This is only allowed if graupel = .T., cloud_scheme = 'morrison' or 'seifert_beheng' and microphysics_ice_phase = .T..
start_ice_microphysics#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: s
Time after which ice microphysicsal processes are calculated.
This requires microphysics_ice_phase = .T..
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Enables/disables entire module.
When set to .T. will turn the entire module off without removing the namelist.
ventilation_effect#
- Fortran Type: L
-
Default: .TRUE.
Enables/disables the effect of ventilation on the evaporation of raindrops.
This parameter is only used if cloud_scheme = 'morrison' or 'seifert_beheng'.
Chemistry Parameters#
bc_cs_b#
- Fortran Type: C*20
-
Default: dirichlet
Bottom boundary condition of the chemical species (cs) concentration. For constant surface flux bc_cs_b must be set to 'neumann'.
Currently 2 choices are available:
-
dirichlet
cs(k=0) = constant = cs_surface;
-
neumann
When a constant surface concentration flux is used (surface_csflux) or emissions_anthropogenic = .T.), bc_cs_b = 'neumann' must be used.
-
bc_cs_l#
- Fortran Type: C*20
-
Default: undefined
Left boundary condition of the chemical species (cs) concentration.
If the value of the parameter is not set by the user, then the value given by the parameter bc_lr will be used. If 'cyclic' has been chosen, parameter bc_cs_r must also be set to 'cyclic'. Horizontal boundary conditions for chemical species can be set independently from horizontal boundary conditions that have been chosen for the remaining model quantities via parameters bc_lr and bc_ns. For instance, you can choose cyclic boundary conditions for chemical species, while the flow field uses non-cyclic conditions, or vice-a-versa.
For nested runs (child domain or offline-nested root domain) boundary values will be set using values from parent domains or from the dynamic driver file, depending on the setting of nesting_offline_chem and nesting_chem.
Currently 3 choices are available:
-
dirichlet
Constant concentration over the entire simulation as given by the initial profiles;
-
neumann
Zero concentration gradient;
-
cyclic
-
bc_cs_n#
- Fortran Type: C*20
-
Default: undefined
North boundary condition of the chemical species (cs) concentration.
If the value of the parameter is not set by the user, then the value of the parameter bc_ns will be used. If 'cyclic' boundary condition has been chosen, then the parameter bc_cs_s must also be set to 'cyclic'. Horizontal boundary conditions for chemical species can be set independently from horizontal boundary conditions that have been chosen for the meteorological quantities via parameters bc_lr and bc_ns. For instance you can choose cyclic boundary conditions for chemical species, while the flow field uses non-cyclic conditions, or vice-a-versa.
For nested runs (child domain or offline-nested root domain) boundary values will be set using values from parent domains or from the dynamic driver file, depending on the setting of nesting_offline_chem and nesting_chem.
Currently 3 choices are available:
-
dirichlet
Constant concentration over the entire simulation as given by the initial profiles;
-
neumann
Zero concentration gradient;
-
cyclic
-
bc_cs_r#
- Fortran Type: C*20
-
Default: undefined
Right boundary condition of the chemical species (cs) concentration.
If the value of the parameter is not set by the user, then the value of the parameter bc_lr will be used. If 'cyclic' boundary condition has been chosen, then the parameter bc_cs_l must also be set to 'cyclic'. Horizontal boundary conditions for chemical species can be set independently from horizontal boundary conditions that have been chosen for the meteorological quantities via parameters bc_lr and bc_ns. For instance, you can choose cyclic boundary conditions for chemical species, while the flow field uses non-cyclic conditions, or vice-a-versa.
For nested runs (child domain or offline-nested root domain) boundary values will be set using values from parent domains or from the dynamic driver file, depending on the setting of nesting_offline_chem and nesting_chem.
Currently 3 choices are available:
-
dirichlet
Constant concentration over the entire simulation as given by the initial profiles;
-
neumann
Zero concentration gradient;
-
cyclic
-
bc_cs_s#
- Fortran Type: C*20
-
Default: undefined
South boundary condition of the chemical species (cs) concentration.
If the value of the paramenter is not set by the user, then the value of the parameter bc_ns will be used. If 'cyclic' boundary condition has been chosen, parameter bc_cs_s must be set to 'cyclic'. Horizontal boundary conditions for chemical species can be set independently from horizontal boundary conditions that have been chosen for the meteorological quantities via parameters bc_lr and bc_ns. For instance, you can choose cyclic boundary conditions for chemical species, while the flow field uses non-cyclic conditions, or vice-a-versa.
For nested runs (child domain or offline-nested root domain) boundary values will be set using values from parent domains or from the dynamic driver file, depending on the setting of nesting_offline_chem and nesting_chem.
Currently 3 choices are available:
-
dirichlet
Constant concentration over the entire simulation as given by the initial profiles;
-
neumann
zero concentration gradient;
-
cyclic
-
bc_cs_t#
- Fortran Type: C*20
-
Default: initial_gradient
Top boundary condition of the chemical species (cs) concentration.
For nested runs (child domain or offline-nested root domain) boundary values will be set using values from parent domains or from the dynamic driver file, depending on the setting of nesting_offline_chem and nesting_chem.
Currently 3 choices are available:
-
dirichlet
Constant concentration over the entire simulation as given by the initial profiles;
-
neumann
zero concentration gradient (cs(k=nz+1) = cs(k=nz));
-
initial_gradient
value of the scalar concentration gradient at the top is calculated from the initial scalar concentration profile (see cs_profile). Using this value (assumed constant during the run) the concentration boundary values are calculated as cs(k=nz+1) = cs(k=nz) + bc_cs_t_val * dzu(nz+1) with bc_cs_t_val being the concentration gradient between levels nzt and nzt+1.
-
call_chem_at_all_substeps#
- Fortran Type: L
-
Default: .FALSE.
Switch, whether chemistry is called (.TRUE.) or not called (.FALSE.) at intermediate substepss of the Runge-Kutta scheme. Recommended is the default (.FALSE.).
Switch, whether chemistry is called at intermediate substeps of the Runge-Kutta scheme. Note that seting this parameter to .TRUE. is only appropriate for test purposes and does not increase the accuracy since the intermediate steps of the Runge-Kutta scheme do not represent a physical state.
chem_gasphase_on#
- Fortran Type: L
-
Default: .TRUE.
Switches the gas-phase chemistry ON (.TRUE.) or OFF (.FALSE.)
When the parameter is set to .TRUE. both gas-phase reactions and transport are activated. If the parameter is switched OFF, no chemical reactions will be calculated, only the transport for all chemical compounds will occur. This is useful for test purposes.
chem_isorropia#
- Fortran Type: L
-
Default: .FALSE.
Activation of ISORROPIA coupling
chem_isorropia_activity_coefficient_method#
- Fortran Type: I
-
Default: 1
Method of activity coefficient calculation
Currently 2 choices are available:
-
0
Calculate activity coefficients during runtime.
-
1
Use precalculated tables for activity coefficients. See ISORROPIA model description
-
chem_isorropia_activity_tolerance#
- Fortran Type: R
-
Default: 0.05
Activity coefficient convergence criterion
Defines the convergence criterion for activity coefficient calculations in ISORROPIA. Any positive floating point is possible. See ISORROPIA model description
chem_isorropia_aerosol_state#
- Fortran Type: R
-
Default: 0.0
Defines the aerosol state
Currently 2 choices are available:
-
0.0
The aerosol can have both solid and liquid phases (deliquescent).
-
1.0
The aerosol is in only liquid state (metastable aerosol). See ISORROPIA model description
-
chem_isorropia_mass_conservation_mode#
- Fortran Type: I
-
Default: 1
Mass conservation enforcement (ISORROPIA II only)
Defines whether ISORROPIA will force conservation of mass up to the precision of the machine. This option is only available for ISORROPIA II.
Currently 2 choices are available:
-
0
Normal calculation mode.
-
1
After calculating the thermodynamic solution, “adjust” concentrations of species so that total conservation of mass is satisfied to machine precision. See ISORROPIA model description
-
chem_isorropia_mdr_weight_method#
- Fortran Type: I
-
Default: 0
Type of weighting algorithm for mutual deliquescence regions (MDR)
Defines the type of weighting algorithm for the solution in Mutual Deliquescence Regions (MDR's).
Currently 3 choices are available:
-
0
MDRs are assumed dry. This is equivalent to the approach used in equilibrium routine SEQUILIB.
-
1
The solution is assumed half dry and half wet throughout the MDR.
-
2
The solution is a relative-humidity weighted mean of the dry and wet solutions. See ISORROPIA model description
-
chem_isorropia_max_activity_sweep#
- Fortran Type: I
-
Default: 4
Maximum activity coefficient sweeps in ISORROPIA
Defines the maximum number of iterations for activity coefficient calculations in ISORROPIA. Any positive integer is possible. See ISORROPIA model description
chem_isorropia_max_iteration#
- Fortran Type: I
-
Default: 100
Maximum number of iterations in ISORROPIA
Defines the maximum number of iterations for all iterative processes in ISORROPIA, except those for activity coefficient calculations (see also chem_isorropia_max_activity_sweep). Any positive integer is possible. See ISORROPIA model description
chem_isorropia_problem_type#
- Fortran Type: R
-
Default: 0.0
Maximum number of iterations in ISORROPIA
Currently 2 choices are available:
-
0.0
Forward problem is solved. In this case, the array of species concentrations contains GAS and AEROSOL concentrations together.
-
1.0
Reverse problem is solved. In this case, the array of species concentrations contains AEROSOL concentrations only. See ISORROPIA model description
-
chem_isorropia_solver_tolerance#
- Fortran Type: R
-
Default: 1e-06
Solver convergence criterion
Defines the convergence criterion for all iterative processes in ISORROPIA, except those for activity coefficient calculations (chem_isorropia_activity_tolerance controls that). Any positive floating point is possible. See ISORROPIA model description
chem_isorropia_root_subdivisions#
- Fortran Type: I
-
Default: 5
Number of subdivisions for root tracking
Defines the number of subdivisions for root tracking in ISORROPIA, when the bisection method is used for numerically solving equilibrium equations. Normally this parameter should not have to change, but is included for completeness. Any positive integer is possible. See ISORROPIA model description
chem_wet_deposition#
- Fortran Type: L
-
Default: .FALSE.
Activation of wet deposition module
Turns on the wet deposition parametrization based on Simpson et al 2012, DOI 10.5194/acp-12-7825-2012.
chem_wet_deposition_update_interval#
- Fortran Type: R
-
Default: 300.0
Interval between update of scavenging rates
Scavenging rates are updated at the start of the model run, and afterwards at every user-defined update interval. The default is 300 seconds, but any positive value can be specified.
chem_wet_deposition_model_override#
- Fortran Type: L
-
Default: .FALSE.
Activation of model override
Allows wet desposition to take place without cloud and precipitation input from the bulk cloud model.
chem_wet_deposition_cloud_level_lower#
- Fortran Type: I
-
Default: 10
Vertical level for lower cloud layer
Defines index of lower cloud layer when chem_wet_deposition_model_override is set to .TRUE..
chem_wet_deposition_cloud_level_upper#
- Fortran Type: I
-
Default: 10
Vertical level for upper cloud layer
Defines index of upper cloud layer when chem_wet_deposition_model_override is set to .TRUE..
chem_wet_deposition_rain_rate#
- Fortran Type: R
-
Default: 1.0
Rain rate in domain
Defines the rain rate of all cells when chem_wet_deposition_model_override is set to .TRUE..
chem_mechanism#
- Fortran Type: C*30
-
Default: phstap
Chemical mechanism to be used for gasphase/aerosol chemistry.
The setting in the namelist must match with the mechanism in the code, i.e. in chem_gasphase_mod.f90. For currently available gas-phase chemistry options and how to access them see the chemistry mechanisms summary For the pollen model currently only one mechanism is available i.e."empol1.0". The "empol1.0" must be selected to run the pollen model.
cs_heights#
- Fortran Type: R (99,100)
-
Default: undefined
Height levels for the initial profile of chemical compounds.
Height levels (in m above the gound) to go with cs_profile in order to define initial profiles of chemical species. The first index refers to the chemical compound, and the second to the height level.
Example: cs_heights(1,:) = 0.0, 5.0, 15.0, 25.0, 35.0, 45.0, 55.0, 65.0, 75.0, 85.0, 95.0, (heights for profile of first chemical species) cs_heights(2,:) = 0.0, 5.0, 15.0, 25.0, 35.0, 45.0, 55.0, 65.0, 75.0, 85.0, 95.0, (heights for profile of second chemical species)
cs_name#
- Fortran Type: C*11 (99)
-
Default: novalue
Names of chemical species where surface concentrations or concentration profiles (cs_profile) are prescribed.
Example: cs_name = 'O3', 'NO', 'NO2', 'CO', 'RCHO', 'PM10', 'PM25', It is not necessary to specify cs_name (and cs_surface and cs_profile/cs_heights-pairs) for all compounds of the chosen chemical mechanism. Names of compounds which do not occur in the mechanism, are ignored.
cs_profile#
- Fortran Type: R (99,100)
-
Default: undefined
Concentration values of chemical species (gases in ppm, and PM compounds in kg m-3) at heights cs_heights.
Example: cs_profile(1,:) = 0.020, 0.023, 0.026, 0.029, 0.032, 0.035, 0.038, 0.041, 0.044, 0.047, 0.050, (Values for initial profile of first species) cs_profile(2,:) = 0.080, 0.073, 0.064, 0.057, 0.050, 0.043, 0.036, 0.029, 0.022, 0.015, 0.007, (Values for initial profile of second species)
The individual chemical species are identified using cs_name. These initial profiles become only effective when 'set_constant_profiles' is set for initializing_actions. 'set_constant_profiles' can be combined with 'read_from_file' if the dynamic input file only contains meteorological variables and constant profiles are only set for chemistry.
cs_surface#
- Fortran Type: R (99)
-
Default: 0.0
Initial surface concentrations of chemical compounds (gases in ppm, particulate matter in kg m-3).
Initial surface concentrations of chemical compounds (gases in ppm, and particulate matter in kg m-3) for the chemical species which are specifield by cs_name.
daytype_mdh#
- Fortran Type: C*80
-
Default: workday
Type of weekday (workday, weekend, holiday)
Type of weekday required for the MDH (MonthDayHour) case of the LOD 1 mode of the emissions module. Possible values are: workday, weekend, holiday. See also time_fac_type.
deposition_dry#
- Fortran Type: L
-
Default: .FALSE.
Switch to turn dry deposition ON (.TRUE.) or OFF (.FALSE.).
Switches the deposition calculation for particles and gases ON (.TRUE.) or OFF (.FALSE.).
ebio_dt#
- Fortran Type: R
-
Default: 0.0
Time interval to call the biogenic VOC emission model.
Time-step of biogenic emission model, that is how often biogenic emission would be updated. The default value is 0. If user does not define ebio_dt, the BEM will use models physical time-step. In case user defines ebio_dt but it is less than model's physical time-step then model will issue a warning and make ebio_dt = dt_timestep.
ebio_ef_pft#
- Fortran Type: R
-
Default: undefined
Emission potentials for each vegetation type (PFT).
ebio_ef_tree#
- Fortran Type: R
-
Default: undefined
Emission potentials for each tree type (single trees)
ebio_emis_name#
- Fortran Type: C*11 (99)
-
Default: novalue
Names of the biogenic volatile organic compounds (BVOCs).
Names of biogenic VOCs. The species names must be available in both BVOC Table 1 as well as in the chemical mechanism. User should follow the naming convention in th given chemistry mechanism. At the moment only CBM4 and SMOG chemistry mechanisms have been implemented in the biogenic voc model.
ebio_max_emis_day#
- Fortran Type: I
-
Default: 200
The day when the emissions are expected to be the maximum, The default is 200 which is 19th/20th of July.
ebio_pft#
- Fortran Type: I
-
Default: undefined
Vegetation types (plant functional types). PALM utilizes ECMWF-IFS classification for vegetation types.
ebio_ppfd_factor#
- Fortran Type: R
-
Default: 202.0
Photosynthetic photo flux density (PPFD) factor .
Photosynthetic photo flux density factor (PPFD). The default value is the photosynthetically active radiation(PAR) within the visible range (400-700nm) absorbed by plants. The PPFD value is obtained by converting solar radiaton in \(W m^{-2}\) incident on a grid box to mol \(m^{-2} s^{-1}\).
ebio_rad_method#
- Fortran Type: I
-
Default: undefined
Radiation method for biogenic model defines as how radiation would be used to calculate bvoc emissions.
Currently 2 choices are available:
-
0
The first method is based on calculating the average shortwave radiation (both direct and diffused components) at each plant canopy box (PCB) considering all radiative transfer processes, including shade and reflections. The RTM calculates this flux density using the absorbed radiation at each PCB and given that albedo is set to zero, please see (Krc et al., 2021).
-
1
The second planned method (= 1), is based on calculating the shade ratio of each PCB and the radiation received for the unshaded part. This method is based on the assumption that the radiation reduces from the top face toward the inner centre of the PCB because the leaves on the top surface receive full radiation and leaves inside the grid cell receive less radiation due to being in the shade of the leaves above. This method is not yet implemented.
-
ebio_soilm_method#
- Fortran Type: C*11
-
Default: undefined
Choose the method for calculation fo soil moisture correction factor.
The BEM offers the choice of algorithm in calculation of soil moisture correction factor.
Currently 2 choices are available:
-
bulk
The default option 'bulk' is the simple method same as implemented in MEGAN (Guenther et al., 2012)
-
weighted
The 'weighted' option is more complex and needs detailed data of soil, wilting point and field capacity.
-
ebio_tree#
- Fortran Type: I
-
Default: undefined
Tree types in the simulation domain. The current PALM database has 86 tree types tree types . The user need to choose tree types from this table.
emis_biogenic#
- Fortran Type: L
-
Default: .FALSE.
Switch to turn the biogenic emissions ON (.TRUE.) or OFF (.FALSE.).
emis_biogenic_lod#
- Fortran Type: I
-
Default: 0
Level of detail i.e. biogenic emission mode.
Two differet level of details have been planned for the BVOC model. Currently on LOD 0 is implemented.
Currently 2 choices are available:
-
0
The currently implemented bvoc model is based on Guenther et al., (1999).
-
1
The Model of Emissions of Gases and Aerosols from Nature (MEGAN) is based on Guenther et al., (2012). Not yet implemented.
-
emis_domestic#
- Fortran Type: L
-
Default: .FALSE.
Activation of domestic emissions module
emis_domestic_base_temperature#
- Fortran Type: R
-
Default: 15.0
Base temperature for determining temperature deficit
The mean domestic indoor temperature to be maintained by heating. The default is 15 C.
emis_domestic_compact_factors#
- Fortran Type: R (6)
-
Default: [0.23, 0.28, 0.28, 0.26, 0.29, 0.29]
Compactness factors for each of the six building types
The compactness factor is an indicator of building density, which is defined for each building type (1-6).
emis_domestic_energy_demands#
- Fortran Type: R (6)
-
Default: [30.0, 100.0, 100.0, 11.0, 89.0, 89.0]
Annual energy demand for each of the six building types
The annual specific energy demand estimates the energy requirement for the building per unit footprint area, which is defined for each building type (1-6).
emis_domestic_heating_degree#
- Fortran Type: R
-
Default: 2100.0
Annual heating degree
The amount of heating required on an annual basis are inferred by the so-called annual heating degree, that is, the cumulative temperature, in degrees, to be heated above ambient temperature to the base temperature throughout the year. A value of 2100 K is presented as default.
emis_domestic_lod#
- Fortran Type: I
-
Default: 2
Level of detail for domestic emissions
Currently 2 choices are available:
-
0
Full parameterization (namelist driven)
-
2
No pararmeterization (input supplied in _domestic file)
-
emis_domestic_species_emission_factors#
- Fortran Type: R
-
Default: undefined
Emission factor for each species defined
Emission factors are to be presented on a name-value pair represented by these two arrays in the _p3d file. Emission factors are expressed in a per terajoule (TJ) of energy consumed, and are to be presented in moles for all gas phase species, and kilograms for particulate species. In theory, the user can define up to 20 species, each represented by a name of up to 64 characters. However, only names that are found in the active chemical mechanism will have their corresponding emission factors extracted and used in the parametrization. Due to the variability species names in active chemical mechanisms, the user must provide all species names and corresponding emission factors.
emis_domestic_species_names#
- Fortran Type: C*64 (99)
-
Default: undefined
Name of emission species
Emission factors are to be presented on a name-value pair represented by these two arrays in the _p3d file. Emission factors are expressed in a per terajoule (TJ) of energy consumed, and are to be presented in moles for all gas phase species, and kilograms for particulate species. In theory, the user can define up to 20 species, each represented by a name of up to 64 characters. However, only names that are found in the active chemical mechanism will have their corresponding emission factors extracted and used in the parametrization. Due to the variability species names in active chemical mechanisms, the user must provide all species names and corresponding emission factors.
emis_domestic_update_interval#
- Fortran Type: R
-
Default: 300.0
Interval between emission source updates
Emission sources at each stack location are updated at the start of the model run, and afterwards at every user-defined update interval. The default is 300 seconds, but any positive value can be specified.
emis_generic#
- Fortran Type: L
-
Default: .FALSE.
Activation of generic emissions module
emis_generic_lod#
- Fortran Type: I
-
Default: 2
Level of detail for generic emissions
For generic emission mode only LOD 2 is implemented
Currently 1 choices are available:
-
2
No pararmeterization (input supplied in _generic file)
-
emiss_factor_main#
- Fortran Type: R
-
Default: undefined
Constant emission scaling factor for MAIM street types used in the PARAMETERIZED mode of chem emission module. The number and the order of the values has to correspond to the names of emission species provided for
surface_csflux_name.
emiss_factor_side#
- Fortran Type: R
-
Default: undefined
Constant emission scaling factor for SIDE (secondary) street types used in the PARAMETERIZED mode of chemistry emission module. The number and the order of the values has to correspond to the names of emission species provided for
surface_csflux_name.
emiss_interpolate#
- Fortran Type: L
-
Default: .FALSE.
Enables temporal interpolation of the emission between 'PIDS_CHEM' emission time-steps.
emiss_lod#
- Fortran Type: I
-
Default: undefined
Level of detail/mode of chemistry emissions.
Currently 3 choices are available:
-
0
(=='PARAMETERIZED'): Parameterized traffic emissions based on street types supplied additionally in the _static input file (e.g from OpenStreetMap). Please note that LOD = 0 is not applicable if the horizontal grid exceeds 10 to 15 m at maximum (depending on the width of the streets in the considered area). In this context, it must also be considered, that streets, which are only one grid point wide in the STATIC file will probably be eliminated in the topography filtering during runtime, i.e. emissions from steets which are only one grid point wide will be supressed.
-
1
(=='DEFAULT'): Input of yearly gridded emissions and MDH courses (see also time_fac_type).
-
2
(=='PRE-PROCESSED'): Input of fully preprocessed emissions at fixed time intervals as given in the _chemistry file which contains the emission fields.
-
emis_pollen#
- Fortran Type: L
-
Default: .FALSE.
Parameter to switch pollen emissions ON (.TRUE.) or OFF (.FALSE.)
This parameter is used to activate pollen model. Default is OFF (.FALSE.). To run the pollen model this parameter must be turned on (.TRUE.)
emis_pt_source#
- Fortran Type: L
-
Default: .FALSE.
Switches the LOD 0 point sources mode ON (.TRUE.) or OFF (.FALSE.)
When set to .TRUE. volume source emissions from point sources can be included in the domain via the namelist (LOD 0). Up to 200 point sources can be included.
emis_pt_source_annual_values#
- Fortran Type: R (99)
-
Default: 0.0
Emissions values for point sources (LOD 0) in mol/m3/year.
Annual volume source emissions of point sources (emiss_lod = 0) in mol/m3/year given at locations defined with emis_pt_source_locations_ijk. Example: For two point sources with emissions of three pollutants (NO2, SO2 and PM10) included in the chemistry mechanism and defined with emis_pt_source_species_names = 'no2', 'so2', 'pm10', the emission example values would be given as emis_pt_source_annual_values(1,:) = 100, 200, 300, emis_pt_source_annual_values(2,:) = 100, 200, 300,
emis_pt_source_k_spread#
- Fortran Type: I
-
Default: 3
Z-distribution of point source emission
This parameter allows the user to spread the point source emission over several vertical levels. It is adviced to spread the point source emission over at least 2 levels, 3 is the default.
Currently 5 choices are available:
-
1
Emissions defined using emis_pt_source_annual_values are spread over one vertical level.
-
2
Emissions defined using emis_pt_source_annual_values are spread over two vertical levels.
-
3
Emissions defined using emis_pt_source_annual_values are spread over three vertical levels.
-
4
Emissions defined using emis_pt_source_annual_values are spread over four vertical levels.
-
5
Emissions defined using emis_pt_source_annual_values are spread over five vertical levels.
-
emis_pt_source_k_weights#
- Fortran Type: R (5)
-
Default: Depends on no weights, equal distribution over defined emission levels
Vertical emission distribution weights
Normalized mass distribution weights for the point source emission levels defined using emis_pt_source_k_spread.
Example: In case the user has chosen 5 vertical levels to spread the point source emission (emis_pt_source_k_spread = 5) of all point sources defined in the domain, the weights could exemplary look like this: emis_pt_source_k_weights = 0.6, 0.2, 0.1, 0.05, 0.05,
emis_pt_source_locations_ijk#
- Fortran Type: I
-
Default: undefined
Point source locations in the model domain
Defines the locations of point source emissions in the model domain following the grid spacing (defined in the Initialization parameters) in the horizontal x- (i) and y-directions (j) and in the vertical z-direction (k). Example: For two point sources, the first one located at i = 5, j = 10 and z = 3 and the second one located at i = 15, j = 10 and z = 5. emis_pt_source_location_ijk(1,:) = 5, 10, 3, emis_pt_source_location_ijk(2,:) = 15, 10, 5,
emis_pt_source_species_names#
- Fortran Type: C*64 (99)
-
Default: undefined
Names of chemical compounds of parameterized point source emissions (emiss_lod = 0).
Names of chemical compounds of parameterized point source emissions. The species name should be available in the chemical mechanism. User should follow the naming convention in th given chemistry mechanism. Names that do not appear in the applied chemistry machanism will be ignored.
Example: emis_pt_source_species_names = 'no2', 'so2', 'pm10',
emis_traffic#
- Fortran Type: L
-
Default: .FALSE.
Activation of traffic emissions module
emis_traffic_lod#
- Fortran Type: I
-
Default: 2
Level of detail for traffic emissions
Currently 1 choices are available:
-
2
No pararmeterization (input supplied in _traffic file)
-
emissions_anthropogenic#
- Fortran Type: L
-
Default: .FALSE.
Switches surface emissions ON (.TRUE.) or OFF (.FALSE.) from anthropogenic sources.
epol_ignore_precip#
- Fortran Type: L
-
Default: .TRUE.
Parameter to witch precipitation OFF (.TRUE.) or include (.FALSE.) in the pollen release processes.
Parameter to switch precipitation in OFF (.TRUE.) or include (.FALSE.). in the pollen release processes. Currently precipitation is turned off in the pollen model code.
epol_ignore_solar#
- Fortran Type: L
-
Default: .TRUE.
Switch to ignore (.TRUE.) or include solar radiation (.FALSE.) in the pollen release prrocesses.
Switch to include or exclude solar radiation in the pollen release processes. Currently solar activiy is turned off that is set to (.TRUE.) in the pollen model code.
epol_model#
- Fortran Type: C*64
-
Default: zink
Selection of the pollen model from the available pollen parameterizations.
This parameter offers the choice of pollen emissin parameterizations. Currently only 'zink' (Zink et al., 2013), is implemented and available. In the future releases of the pollen model, 'hlebig' (Helbig et al., 2004) , and 'sofiev', (Sofiev et al., 2013) , would be included.
epol_pool_reset_hour#
- Fortran Type: I
-
Default: 0
Parameter to reset/replenish the pollen pool
The time in UTC when the pollen emission pool is reset and replenished. In most of the cases, it is 00:00 hours local time.
epol_seasonal_factors#
- Fortran Type: R
-
Default: 0.0
Seasonal description of pollen species.
Seasonal description of pollen species. The parameter varies between 0 and 1. This parameter describes start, end and peak of the pollen season. A value of 0 means no pollen emission season, and a value close to 1 indicates peak of pollen emission season.
epol_specs_names#
- Fortran Type: C*64
-
Default: no_value
pollen emitting plant species names, 'POL_BETU',(Birch tree),'POL_ALNU' (Alder tree), 'POL_POAC', (Grasses) and 'POL_AMBR', (Ambrosia/Ragweed)
Names of the pollen emitting plant species. Currently thee species are available, namely, birch (Betula), Alder, and grasses (Poaceae). Due to reasons for algorithm testing and authentication, parameterization for Ambrosia has not not been activated as yet. Ambrosia will be availabe in the next release of the pollen model.
epol_tke_scheme#
- Fortran Type: C*64
-
Default: default
Parameterization to estimate TKE. Currently three TKE schemes are available namely, 'default' 'dynamic', and 'adhoc'
Parameterised turbulence kinetic energy(TKE) for pollen emission processes. Three tke schemes have been implemented namely, 'default', 'dynamic' and 'adhoc'.
epol_tke_sgs_fraction#
- Fortran Type: R
-
Default: 0.1
Fraction to calculatte TKE for the 'default' TKE scheme
The TKE fraction is required to calculate TKE for the 'default' TKE scheme 'epol_tke_scheme'. The value may vary between 0 and 1. The greater the fraction, the higher the magnitude of the TKE.
epol_tree_specs#
- Fortran Type: I
-
Default: undefined
Pollen emitting tree types. Currently two tree and one grass pollen have been implemented, these are, Birch (POL_BETU) with tree id 7, Alder 'POL_ALNU' and grass 'POL_POAC' with vegetation id 3. The index for Alder is not available since Alder is not provided in the tree data base
Pollen emitting tree types. The current PALM database has 86 tree types. The EMPOL model calculates pollen from Birch (betula) and Alder trees. The user can choose other pollen emitting trees also from this database, however, the user need to provide the pollen attributes for the desired tree pollen.
epol_tuning_factors#
- Fortran Type: R
-
Default: 1.0
Tunning factor to adjust maximum possible release of pollen per day per squared meter from a pollen species.
This parameter is used to tune the maximum pollen release per day per squared meter of a particular pollen species based on the observations. The value must be greater than 0. There is no upper bound for this parameter, however, in most of the cases it will remain less than 10.
epol_update_interval#
- Fortran Type: R
-
Default: 300.0
Time interval to update the pollen model.
Time-step of pollen model that is how often the pollen model would be updated. In case user defined update interval is less than model's physical time-step, then the pollen model will issue a warning and set the epol_tuning_factors equals to dt_timestep.
epol_vegetation_specs#
- Fortran Type: I
-
Default: undefined
Pollen emitting vegetation. Currently only grasses (Poaceae) vegetation type is available.
Vegetation types. PALM utilizes ECMWF-IFS classification for vegetation types. The EMPOL model calculates pollen from grasses(poaceae). The user can also choose other pollen emitting vegetation types from this database, however, the user need to provide the pollen attributes for the new vegetation type(s).
icntrl#
- Fortran Type: I (20)
-
Default: 0
Integer steering parameters for the chemistry solver from KPP.
In order to offer more control over the integrator, the KPP-generated integrator subroutine (e.g. SUBROUTINE rosenbrock in chem_gasphase_mod.f90) provides the optional input parameters ICNTRL_U and RCNTRL_U. Each of them is an array of 20 elements that allow the fine-tuning of the integrator, e.g. by setting a particular integrator method, tolerances, minimum and maximum step sizes, etc. Note: For input parameters which are not specified in the namelist the default values of the corresponding variables are used.
ICNTRL(1) = 1: F = F(y) Independent of T (AUTONOMOUS), = 0: F = F(t,y) Depends on T (NON-AUTONOMOUS) ICNTRL(2) = 0: abstol, reltol are N-dimensional vectors, = 1: Abstol, Reltol are scalars ICNTRL(3) -> selection of a particular Rosenbrock method 0 : Rodas3 (Default from KPP) 1 : Ros2 (Simplest Rosenbrock solver, will also do) 2 : Ros3 3 : Ros4 4 : Rodas3 5 : Rodas4 ICNTRL(4) -> maximum number of integration steps; For ICNTRL(4) =0) the default value of 100000 is used. ... See (KPP the Kinetic PreProcessor)
Example (recommended values for PALM): icntrl(3) = 1, ! ros2 (a bit less time consuming than the Rodas3 solver recommended by KPP) icntrl(4) = 500, ! max. number of chem-substeps
main_street_id#
- Fortran Type: I
-
Default: 0
Parameter for emiss_lod = 0 parameterized traffic emissions based on OpenStreetMap.
Index for identifying main streets with high traffic emission following street type classes from OpenStreetMap. This parameter is only required for emiss_lod = 0. main_street_id specifies the starting index for the main streets, i.e. main streets are streets with main_street_id =< street_type < max_street_id. The recommend value is for main_street_id is 11.
street_type must be included in the static file. OpenStreetMap street types are mapped to values for street_type according to the surface classifications given in the description of the static file.
max_street_id#
- Fortran Type: I
-
Default: 0
Parameter for emiss_lod = 0 parameterized traffic emissions based on OpenStreetMap.
Index for identifying the maximum street type class from OpenStreetMap which is considered as a main street. This parameter is only required for emiss_lod = 0. max_street_id specifies the end index for the main streets, i.e. main streets are streets with main_street_id =< street_type < max_street_id. The recommended (and maximum possible) value for max_street_id is 19.
street_type must be included in the static file. OpenStreetMap street types are mapped to values for street_type according to the surface classifications given in the description of the static file.
mode_emis#
- Fortran Type: C*80
-
Default: PARAMETERIZED
Depricated, please use emiss_lod instead,
Mode of chemistry emissions (Depricated), use emiss_lod instead. Possible values are (the values have to be in capital letters): 'DEFAULT' Input of yearly gridded emissions and MDH courses (see also time_fac_type). 'PRE-PROCESSED' Input of fully preprocessed emissions at fixed time intervals as given in the _chemistry file which contains the emission fields.
'PARAMETERIZED' Parameterized traffic emissions based on street types supplied additionally in the _static input file (e.g from OpenStreetMap).
nesting_chem#
- Fortran Type: L
-
Default: .TRUE.
Switch for self-nesting for the chemistry model.
Parameter to switch off offline-nesting for chemistry variables. If nesting_chem = .F. , the boundary conditions for chemistry variables of nest are set according to the setting of bc_cs_b, bc_cs_t, bc_cs_l, bc_cs_r, bc_cs_n, and bc_cs_s.
nesting_offline_chem#
- Fortran Type: L
-
Default: .TRUE.
Switch for offline nesting for the chemistry model.
Parameter to switch off offline-nesting for chemistry variables. If nesting_offline_chem = .F., the boundary conditions for chemistry variables are defined according to the setting of bc_cs_b, bc_cs_t, bc_cs_l, bc_cs_r, bc_cs_n, and bc_cs_s. With nesting_offline_chem = .T., a _dynamic file, which includes at least boundary conditions for some relevant chemistry compounds must be available.
photolysis_scheme#
- Fortran Type: C*10
-
Default: simple
Photolysis Scheme
Photolysis scheme for photochemical reactions.
Currently 2 choices are available:
-
constant
'constant' uses photolysis frequencies at a zenith angle of 45 degrees.
-
simple
'simple' describes parameterized time-dependent photolysis frequencies as supplied with MCM (http://mcm.leeds.ac.uk/MCM, Saunders et al. (2003)). As the solar zenith angle must be available for this scheme, at least the simple clear sky radiation scheme must be active. The 'simple' photolysis scheme also includes shading effect.
-
photolysis_shading#
- Fortran Type: L
-
Default: .FALSE.
Switch to turn ON (.TRUE.) or OFF (.FALSE.) shading effects due to buildings on photolysis frequencies
For photolysis_shading = .T., shading effects of buildings on photolysis frequencies are accounted for depending on buildung heights and solar elevation.
radiation_volumetric_flux = .T. in the radiation namelist section (&radiation_parameters) is required for photolysis_shading = .T.
For photolysis_shading = .T., there is also a simple parameterisation of cloud effects on photolysis whenever the incoming solar radiation rad_sw_in_dir(j,i) + rad_sw_in_diff(j,i) is deviation from the clear sky incoming radiation due to the presence of clouds.
rcntrl#
- Fortran Type: R (20)
-
Default: 0.0
Real type steering parameters for the chemistry solver from KPP.
Steering of the chemistry solver. Note: For input parameters which are not specified in the namelist the default values of the corresponding variables are used.
RCNTRL(1) -> Hmin, lower bound for the integration step size. It is strongly recommended to keep Hmin = ZERO RCNTRL(2) -> Hmax, upper bound for the integration step size RCNTRL(3) -> Hstart, starting value for the integration step size RCNTRL(4) -> Facmin (lower bound on step decrease factor, default=0.2) RCNTRL(5) -> Facmax (upper bound on step increase factor, default=6) RCNTRL(6) -> Facrej (step decrease factor after multiple rejections) RCNTRL(7) -> Facsafe (by which the new step is slightly smaller than the predicted value, default=0.9). ... See (KPP the Kinetic PreProcessor)
Example (recommended values for PALM): rcntrl(3) = 0.1, ! Starting time step of the chemistry solver hstart in sec. ! Setting of hstart can result in savings of computatinal time of 30% and more.
side_street_id#
- Fortran Type: I
-
Default: 0
Parameter for emiss_lod = 0, to identify side streets for parameterized traffic emissions based on OpenStreetMap.
Index for identifying side streets with high traffic emission following street type classes from OpenStreetMap. This parameter is only required for (emiss_lod = 0. side_street_id specifies the starting index for the side streets, i.e. side streets are streets with moderate emissions. side_street_id =< street_type < main_street_id. The recommend value is for side_street_id is 8.
street_type must be included in the static file. OpenStreetMap street types are mapped to values for street_type according to the surface classifications given in the description of the static file.
surface_csflux#
- Fortran Type: R (99)
-
Default: 0.0
Surface emissions of chemical compounds for parameterized emissions (emiss_lod = 0).
Values of surface fluxes of chemistry emissions to be used for emiss_lod = 0 (PARAMETERIZED mode). This parameter is only required for LOD 0!
The number and the order of the values has to correspond to the names of the emission species provided to surface_csflux_name](#chemistry_parameters--surface_csflux_name).
Units of the input values differ between gases and PMs. For gases, surface_csflux must be provided in micromoles/m2*d, while for PMs, input emission values have to be in kg/m2*d.
It is not necessary to specify surface_csflux_name and surface_csflux for all compounds of the chosen chemical mechanism. Names of compounds which do not occur in the mechanism are ignored.
surface_csflux_name#
- Fortran Type: C*11 (99)
-
Default: novalue
Names of chemical compounds of surface emissionsfor parameterized emissions (emiss_lod = 0).
Values of surface fluxes of chemistry emissions to be used for emiss_lod = 0 (PARAMETERIZED mode). This parameter is only required for LOD 0!
The number and the order of the values of surface_csflux](#chemistry_parameters--surface_csflux) has to correspond to the order specified by surface_csflux_name.
Example: surface_csflux_name = 'NO', 'NO2', 'CO', 'RCHO', 'PM10', 'PM25', It is not necessary to specify surface_csflux_name and surface_csflux for all compounds of the chosen chemical mechanism. Names of compounds which do not occur in the mechanism are ignored.
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Local namelist parameter to switch off the module
Allows to switch off the chemistry module entirely for switch_off_module = .T.
time_fac_type#
- Fortran Type: C*80
-
Default: MDH
Type of time treatment for LOD 1 emissions
Type of time treatment for emiss_lod = 1 mode of the chem_emission module. Possible values are: HOUR or MDH (MonthDayHour).
Dust Emission and Transport Parameters#
alpha_imp#
- Fortran Type: R
-
Default: 50.0
Parameter that modifies the magnitude of the impaction efficiency.
If deposition_scheme = 'Z01', the impaction efficiency is calculated by a power function that depends on the Stockes number and alpha_imp. Equation (7c) of Zhang et al. (2001) is used. Depending on the land use category that is assumed, alpha_imp can be different. The default value is for deserts.
bc_dm_b#
- Fortran Type: C*20
-
Default: neumann
The bottom boundary condition of the dust mass concentrations.
The bottom boundary condition for all dust mass concentrations (for each dust size bin). Because surface emissions are always active, bc_dm_b = 'neumann' is always required.
Currently 1 choices are available:
-
neumann
Zero concentration gradient.
-
bc_dm_l#
- Fortran Type: C*20
-
Default: cyclic
The left boundary condition of the dust mass concentrations.
The left boundary condition for all dust mass concentrations (for each dust size bin). So far, only cyclic boundary conditions along the horizontal directions are allowed.
Currently 1 choices are available:
-
cyclic
Cyclic along
x. For this value, bc_dm_r = 'cyclic' is required, too.
-
bc_dm_n#
- Fortran Type: C*20
-
Default: cyclic
The north boundary condition of the dust mass concentrations.
The north boundary condition for all dust mass concentrations (for each dust size bin). So far, only cyclic boundary conditions along the horizontal directions are allowed.
Currently 1 choices are available:
-
cyclic
Cyclic along
y. For this value, bc_dm_s = 'cyclic' is required, too.
-
bc_dm_r#
- Fortran Type: C*20
-
Default: cyclic
The right boundary condition of the dust mass concentrations.
The right boundary condition for all dust mass concentrations (for each dust size bin). So far, only cyclic boundary conditions along the horizontal directions are allowed.
Currently 1 choices are available:
-
cyclic
Cyclic along
x. For this value, bc_dm_l = 'cyclic' is required, too.
-
bc_dm_s#
- Fortran Type: C*20
-
Default: cyclic
The south boundary condition of the dust mass concentrations.
The south boundary condition for all dust mass concentrations (for each dust size bin). So far, only cyclic boundary conditions along the horizontal directions are allowed.
Currently 1 choices are available:
-
cyclic
Cyclic along
y. For this value, bc_dm_n = 'cyclic' is required, too.
-
bc_dm_t#
- Fortran Type: C*20
-
Default: neumann
The top boundary condition of the dust mass concentrations.
The top boundary condition for all dust mass concentrations (for each dust size bin).
Currently 2 choices are available:
-
dirichlet
Constant concentration over the entire simulation.
-
neumann
Zero concentration gradient.
-
bin_mass_fraction_ssc#
- Fortran Type: R (20)
-
Default: [1.0, 0.25, 0.25, 0.25, 0.25, 0.0205, 0.041, 0.0359, 0.3897, 0.5128]
Bin-specific mass fraction of corresponding soil separate class.
With this parameter, the mass fraction of every salation size bin is set with respect to a specific soil separate class (clay, silt or sand). For example, the default setting assumes 1 clay, 4 silt, and 5 sand bins. Because there is only one clay bin the mass fraction is 1.0. The silt bins have uniform mass fractions of 0.25 and the 5 sand bins have fractions that increase from 0.0205 to 0.5128. All mass fractions of each soil separate class should amount to 1 (100%). Up to 20 values can be specified.
Note:
One clay bin must at least be specified. Otherwise, the sandblasting efficiency (see Marticorena et al. (1995) equation (47)) can not be calculated.
brownian_diffusion_coefficient#
- Fortran Type: R
-
Default: 0.54
Exponent that scales Brownian diffusion.
If deposition_scheme = 'Z01', the particle collection efficiency from Brwonian diffusion is calculated by the Schmidt number to the power of - brownian_diffusion_coefficient. Equation (6) of Zhang et al. (2001) is used. Depending on the land use category that is assumed, brownian_diffusion_coefficient can be different. The default value is for deserts.
deposition_scheme#
- Fortran Type: C*4
-
Default: Z01
Method to solve the dry deposition velocity for dust particels over the ground.
The default method is described by Zhang et al. (2001). This parameterization calculates particle dry deposition velocities as a function of particle size and density as well as relevant meteorological variables. It includes deposition processes, such as, turbulent transfer, Brownian diffusion, impaction, interception, gravitational settling and particle rebound. It is widely used in numerical codes, e.g. in WRF-Chem (Zeng et al. (2020)). So far, this is the only implemented method.
Currently 1 choices are available:
-
Z01
See Zhang et al. (2001).
-
det_start_time#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: s
Time after which DET is switched on.
Can be used to allow the LES model to develop turbulence before dust physics are switched on.
diameter_dust#
- Fortran Type: R (10)
- Default: [1.46e-06, 2.8e-06, 4.8e-06, 9e-06, 1.6e-05]
-
SI-Unit: m
Effective diameter of a dust size bin.
By default, effective diameters for 5 dust particle size bins are given. They are mainly used to distribute the calculated bulk dust emission flux to different bins, to calculate the settling velocities, and to calculate dry deposition. The highest given diameter must be equal or less than 63.0E-6. Particle diameters larger than that are considered as sand for which the dust emission and transport module is not designed. Up to 10 values can be specified.
diameter_saltation#
- Fortran Type: R (20)
- Default: [1.42e-06, 8e-06, 2e-05, 3.2e-05, 4.4e-05, 7e-05, 0.00013, 0.0002, 0.00062, 0.0015]
-
SI-Unit: m
Effective diameter of a saltation size bin.
By default, effective diameters for 10 saltation particle size bins are given. They are mainly used to calculate the bin-specific threshold friction velocities above which saltation occurs and to calculate the weighting factors that determine the contribution of the specific bin to the total horizontal saltation flux. Up to 20 values can be specified.
lower_bound_diameter#
- Fortran Type: R (10)
- Default: [2e-07, 2e-06, 3.6e-06, 6e-06, 1.2e-05]
-
SI-Unit: m
Lowest diameter of a certain dust size bin.
This parameter represents the minimum effective diameter of a certain dust size bin. The maximum is characterized by upper_bound_diameter. It is used to calculate the normalized volume distributions of each dust size bin. Up to 10 values can be specified.
mass_fraction_ssc#
- Fortran Type: R (20)
-
Default: [0.03, 0.05, 0.05, 0.05, 0.05, 0.92, 0.92, 0.92, 0.92, 0.92]
Mass fraction of soil separate class.
With this parameter, the mass fractions of every soil separate class (clay, silt or sand) are set. For example, the default setting assumes 1 clay, 4 silt, and 5 sand bins. All clay, silt, and sand bins contirbute to 3%, 5%, and 92%, respectively, to the soil composition. These values should amount to 1 (100%) and they are based on mass fractions of the soil type sand of the STATSGO-FAO database (Pérez, et al. (2011). Up to 20 values can be specified.
Note:
One clay bin must at least be specified. Otherwise, the sandblasting efficiency (see Marticorena et al. (1995) equation (47)) can not be calculated.
n_dust_bins#
- Fortran Type: I
-
Default: 5
Number of dust particle bins.
By default, 5 dust size bins are specified among which the calculated bulk dust emission flux is distributed and for which the prognostic equations are solved.
n_saltation_bins#
- Fortran Type: I
-
Default: 10
Number of saltation particle bins.
By default, 10 saltation bins are specified for which particle-size-dependent horizontal saltation fluxes are calculated to finally determine the vertical bulk dust emission flux.
particle_density_dust#
- Fortran Type: R (10)
- Default: [2500.0, 2650.0, 2650.0, 2650.0, 2650.0]
-
SI-Unit: kg m-3
Particle density of a dust size bin.
By default, 5 dust bins and their particle densities are specified. Up to 10 can be given.
particle_density_saltation#
- Fortran Type: R (20)
- Default: [2500.0, 2650.0, 2650.0, 2650.0, 2650.0, 2650.0, 2650.0, 2650.0, 2650.0, 2650.0]
-
SI-Unit: kg m-3
Particle density of a saltation size bin.
By default, 10 saltation bins and their particle densities are specified. Up to 20 can be given.
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Enables/Disables entire module.
When set to .TRUE., the entire module is switched off without the requirement to remove the namelist from the parameter file.
upper_bound_diameter#
- Fortran Type: R (5)
- Default: [2e-06, 3.6e-06, 6e-06, 1.2e-05, 2e-05]
-
SI-Unit: m
Highest diameter of a certain dust size bin.
This parameter represents the maximum effective diameter of a certain dust size bin. The minimum is characterized by lower_bound_diameter. It is used to calculate the normalized volume distributions of each dust size bin.
FASTv8 Coupler Parameters#
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Flag to switch on/off the this module.
If switch_off_module = .T., this module is not activated even if the namelist is present in the parameter file.
nturbines#
- Fortran Type: I
-
Default: 0
Number of individual turbines.
The number of turbines set in order to tell PALM, with how many FASTv8 instances it is supposed to establish a TCP/IP connection.
time_turbine_on#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: s
Time delay relative to the PALM simulation time after which the FASTv8 instance coupling is started.
This can be set to a value greater than 0.0, in order to delay the activation of all coupled turbines.
fast_n_blades_max#
- Fortran Type: I
-
Default: 0
Maximum number of blades that an individual FASTv8 instance is allowed to communicate to PALM.
Please set this to the highest number of blades, a turbine has in your simulation setup.
fast_n_blade_elem_max#
- Fortran Type: I
-
Default: 0
Maximum number of blade elements that an individual FASTv8 server instance is allowed to communicate to PALM.
Please set this to the highest number of blade elements, a turbine has in your simulation setup.
fast_host_addr_default#
- Fortran Type: C*15
-
Default: 127.0.0.1
Default IPv4 address that all FASTv8 server instances are expected to run on.
PALM uses this address in order to establish communication with all configured FASTv8 instances. This can be set to any valid IPv4 address. This value is only used in case fast_host_addr is not set for a specific turbine.
fast_host_addr#
- Fortran Type: C*15 (300)
-
Default: Value of fast_host_addr_default
IPv4 address that a specific FASTv8 server instance is expected to run on.
PALM uses this address in order to establish communication with each of the configured FASTv8 instances. For up to 300 FASTv8 server instances, a individual IPv4 address can be configured.
fast_host_port#
- Fortran Type: C*5 (300)
-
Default: undefined
Network port number on which a specific FASTv8 server instance is expected to listen for incoming network connections.
PALM uses this port number in order to establish communication with each of the configured FASTv8 instances. For up to 300 FASTv8 server instances, a individual IPv4 address can be configured.
palm_tower_ref_pos_x#
- Fortran Type: R (300)
- Default: undefined
-
SI-Unit: m
Turbine tower base reference position in X direction inside the PALM domain for each turbine.
This must be set to a positiv value that falls inside the configured PALM model domain. Note, that as the the wind velocities PALM is communicating to FASTv8 are retrieved at 2x the respective rotor diameter upstream from the turbine position, this value must at least be set so that is has a distance from the left domain boundary of more that 2x the rotor diameter.
palm_tower_ref_pos_y#
- Fortran Type: R (300)
- Default: undefined
-
SI-Unit: m
Turbine tower base reference position in Y direction inside the PALM domain for each turbine.
This must be set to a positiv value that falls inside the configured PALM model domain.
palm_tower_ref_pos_z#
- Fortran Type: R (300)
- Default: 0.0
-
SI-Unit: m
Turbine tower base reference position in Z direction inside the PALM domain for each turbine.
This must be set to a positiv value that falls inside the configured PALM model domain.
reg_fac#
- Fortran Type: R (300)
- Default: 2.0
-
SI-Unit: m
Regularization factor for force distribution for each turbine.
fbox_fac#
- Fortran Type: R (300)
-
Default: 1.5
Force distribution cut-off factor for each turbine.
Edge length of the box around each turbine in which forces are distributed (edge length = edgelen_fac * turbine radius). Around every point of attack there should be the at least the distance of 3 * grid spacing * reg_fac for force distribution available.
dtow#
- Fortran Type: R (300)
-
Default: 0.0
Turbine tower diameter for each turbine.
htow#
- Fortran Type: R (300)
-
Default: 0.0
Turbine tower height for each turbine.
turb_C_d_tow#
- Fortran Type: R (300)
-
Default: 1.2
Drag coefficient for each turbine tower.
Indoor Parameters#
high_vent_end_industry_building#
- Fortran Type: C*23
-
Default: 2019-06-21 22:00:00 +00
End date and time of high ventilation load for industry buildings.
The default is set to 21st of June 2019 at 22UTC. Please see also explanations given for origin_date_time. The given date should correspond to origin_date_time.
high_vent_end_office_building#
- Fortran Type: C*23
-
Default: 2019-06-21 18:00:00 +00
End date and time of high ventilation load for office buildings.
The default is set to 21st of June 2019 at 22UTC. Please see also explanations given for origin_date_time. The given date should correspond to origin_date_time.
high_vent_end_residential_building#
- Fortran Type: C*23
-
Default: 2019-06-21 18:00:00 +00
End date and time of high ventilation load for residential buildings.
The default is set to 21st of June 2019 at 22UTC. Please see also explanations given for origin_date_time. The given date should correspond to origin_date_time.
high_vent_start_industry_building#
- Fortran Type: C*23
-
Default: 2019-06-21 06:00:00 +00
Start date and time of high ventilation load for industry buildings.
The default is set to 21st of June 2019 at 22UTC. Please see also explanations given for origin_date_time. The given date should correspond to origin_date_time.
high_vent_start_office_building#
- Fortran Type: C*23
-
Default: 2019-06-21 08:00:00 +00
Start date and time of high ventilation load for office buildings.
The default is set to 21st of June 2019 at 22UTC. Please see also explanations given for origin_date_time. The given date should correspond to origin_date_time.
high_vent_start_residential_building#
- Fortran Type: C*23
-
Default: 2019-06-21 08:00:00 +00
Start date and time of high ventilation load for residential buildings.
The default is set to 21st of June 2019 at 22UTC. Please see also explanations given for origin_date_time. The given date should correspond to origin_date_time.
indoor_during_spinup#
- Fortran Type: L
-
Default: .FALSE.
Switch on the indoor-climate model during wall/soil spinup.
initial_indoor_temperature#
- Fortran Type: R
- Default: 293.15
-
SI-Unit: K
Initial indoor temperature.
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Switch to turn off the module.
If .T., the indoor-climate module is not activated even if the namelist is present in the parameter file.
warm_season_start#
- Fortran Type: I
-
Default: 80
Begin of warm season given in days of year.
warm_season_end#
- Fortran Type: I
-
Default: 264
End of warm season given in days of year.
Land Surface Parameters#
aero_resist_kray#
- Fortran Type: L
-
Default: .TRUE.
Flag for switching on the parameterization of the aerodynamic resistance after Krayenhoff & Voogt (2007).
For homogeneous surface elements, Monin-Obukhov theory is used to calculate the aerodynamic resistance. As the involved stability correction does not apply for vertically-oriented surfaces, the aerodynamic resistance parameterization after Krayenhoff & Voogt (2007) is employed if this parameter is set to .T..
alpha_vangenuchten#
- Fortran Type: R
-
Default: Depends on soil_type
Coefficient alpha in the soil hydraulic conductivity parametrization
Value of the coefficient alpha for the calculation of the hydraulic conductivity of soil in the parametrization after Van Genuchten (1980). This parameter is considered to be equal for all soil layers.
c_surface#
- Fortran Type: R
- Default: Depends on vegetation_type
-
SI-Unit: J m\(^{-2}\) K\(^{-1}\)
Heat capacity of the surface (or skin layer)
Heat capacity of the surface (or skin layer) per unit of area in case of vegetated surfaces. Note that this parameter does not apply in case of water_type and water_type surfaces.
canopy_resistance_coefficient#
- Fortran Type: R
- Default: Depends on vegetation_type
-
SI-Unit: hPa\(^{-1}\)
Coefficient for calculating the canopy water vapor pressure deficit
Only relevant for high vegetation. A coefficient for the dependence of the canopy resistance on water vapor pressure deficit.
conserve_water_content#
- Fortran Type: L
-
Default: .TRUE.
Flag parameter for setting the bottom boundary condition of the soil model.
This flag parameter sets the bottom boundary condition for the soil model. The user can choose between the following options:
-
.T.: Closed bottom, considering bedrock below the soil model. No drainage is possible from the lowest soil model so that conservation of water in the model is guaranteed. Note that this conservation might be compromized by the numerical schemes used in the atmosphere.
-
.F.: Open bottom, considering free drainage. Water can leave the soil model via drainage. There is no conservation of water in the model.
-
constant_roughness#
- Fortran Type: L
-
Default: .TRUE.
Flag parameter for switching on a Charnock parameterization to account for sea surface waves.
This parameter only applied to water_surfaces. If set to .F., the aerodynamic roughness length is allowed to vary in space and time in order to account for increasing surface roughness due to sea surface waves. The roughness length then depends on the near-surface wind speed, following the parameterizations after Charnock (1955) and Beljaars (1994). Note that this parameterization assumes a infinite sea surface that allows for the build-up of significant waves. This option should only be used with special care and only if the waves height is much below the vertical grid spacing (i.e. the waves should be a sub-grid scale feature). Further, note that the computation of the MOST relations requires that the resulting roughness length is smaller than the height of the first prognostic grid level above the surface. If this is not fulfilled, the simulation will be terminated by default. In order to avoid this, set allow_roughness_limitation = .T. to limit the roughness length accordingly.
deep_soil_temperature#
- Fortran Type: R
- Default: undefined
-
SI-Unit: K
Bottom boundary condition temperature for the soil model.
The deep soil temperature is used as bottom boundary condition for the heat diffusion in the soil model. Note, in case that the soil temperature is provided by the dynamic driver file, the deep soil temperature will be taken from the deepest level therein.
dz_soil#
- Fortran Type: R (20)
- Default: [0.01, 0.02, 0.04, 0.06, 0.14, 0.26, 0.54, 1.86]
-
SI-Unit: m
Thickness of the individual soil layers.
Thickness of the individual soil layers. The default configuration considers 8 soil layers. The user can specify up to 20 layers, though. Note that the model operates with negative values in the vertical direction downwards, which will then appear in the output files. The default soil layer configuration is given below.
Layer # dz_soil Depth (m) (m) Layer 1 0.01 0.00 - 0.01 Layer 2 0.02 0.01 - 0.03 Layer 3 0.04 0.03 - 0.07 Layer 4 0.06 0.07 - 0.13 Layer 5 0.14 0.13 - 0.27 Layer 6 0.26 0.27 - 0.51 Layer 7 0.54 0.51 - 1.05 Layer 8 1.86 1.05 - 2.91 The setting of dz_soil can be overwritten if respective data is provided by the dynamic driver file.
field_capacity#
- Fortran Type: R
- Default: Depends on soil_type
-
SI-Unit: m\(^3\) m\(^{-3}\)
Soil moisture at field capacity
Soil moisture at field capacity. This parameter is considered to be equal for all soil layers.
f_shortwave_incoming#
- Fortran Type: R
-
Default: Depends on soil_type
Fraction of the net shortwave radiation that is transmitted directly to the top soil layer.
Fraction of the net shortwave radiation that is transmitted directly to the top soil layer (range of 0-1). The remaining fraction of the shortwave radiation is absorbed by the surface (or skin layer). This parameter has not been fully implemented yet and is fixed to 0.
hydraulic_conductivity#
- Fortran Type: R
- Default: Depends on soil_type
-
SI-Unit: m s\(^{-1}\)
Hydraulic conductivity of the soil at saturation.
Hydraulic conductivity of the soil at saturation. This parameter is considered to be equal for all soil layers.
lambda_surface_stable#
- Fortran Type: R
- Default: Depends on vegetation_type
-
SI-Unit: W m\(^{-2}\) K\(^{-1}\)
Heat conductivity between atmosphere and soil (stable stratification).
lambda_surface_unstable#
- Fortran Type: R
- Default: Depends on vegetation_type
-
SI-Unit: W m\(^{-2}\) K\(^{-1}\)
Heat conductivity between atmosphere and soil (unstable stratification).
leaf_area_index#
- Fortran Type: R
- Default: Depends on vegetation_type, static driver file
-
SI-Unit: m\(^2\) m\(^{-2}\)
Leaf area index of the vegetation canopy.
Leaf area index of the vegetation canopy. The setting of leaf_area_index can be overwritten if respective data is provided by the static driver file.
l_vangenuchten#
- Fortran Type: R
-
Default: Depends on soil_type
Coefficient
lin the soil hydraulic conductivity parametrization.Coefficient
lin the soil hydraulic conductivity of the soil in the parameterization after Van Genuchten (1980).
min_canopy_resistance#
- Fortran Type: R
- Default: Depends on vegetation_type
-
SI-Unit: s m\(^{-1}\)
Minimum canopy (i.e. the stomatal) resistance to evaporation.
min_soil_resistance#
- Fortran Type: R
- Default: 50.0
-
SI-Unit: s m\(^{-1}\)
Minimum soil resistance of the uppermost soil layer to evaporation.
n_vangenuchten#
- Fortran Type: R
-
Default: Depends on soil_type
Coefficient
nin the soil hydraulic conductivity of the soil in the parameterization after Van Genuchten (1980).
pavement_depth_level#
- Fortran Type: I
-
Default: Depends on pavement_type
Number of pavement levels.
Extent of the pavement layer in grid levels below the surface. Each layer can only be 100% pavement or 100% soil.
pavement_heat_capacity#
- Fortran Type: R
- Default: Depends on pavement_type
-
SI-Unit: J K\(^{-1}\) m\(^{-3}\)
Heat capacity of the pavement layer.
If this parameter is set, the value is assumed for all pavement layers.
pavement_heat_conduct#
- Fortran Type: R
- Default: Depends on pavement_type
-
SI-Unit: W m\(^{-1}\) K\(^{-1}\)
Thermal conductivity of the pavement layer.
If this parameter is set, the value is assumed for all pavement layers.
pavement_type#
- Fortran Type: I
-
Default: 1
Pavement type according to internal classification
Pavement type according to PALM's internal classification (see list below) and which loosely follows the OpenStreetMap classification. Note that the default pavement types (i.e. pavement_type /= 0) is only allowed in combination with the default soil layer depth configuration (see dz_soil). The setting of pavement_type can be overwritten if respective data is provided by the static driver file.
pavement_type Description Notes 1 Asphalt/concrete mix 2 Asphalt (asphalt concrete) 3 Concrete (Portland concrete) 4 Sett 5 Paving stones 6 Cobblestone 7 Metal 8 Wood 9 Gravel 10 Fine gravel 11 Pebblestone 12 Woodchips 13 Tartan (sports) 14 Artifical turf (sports) 15 Clay (sports) Setting of the pavement type implies the setting of the following parameters:
pavement_type z0_pavement z0h_pavement albedo_type emissivity pavement_depth_level (m) (m) 1 5.0E-2 5.0E-4 18 0.93 7 2 5.0E-2 5.0E-4 19 0.95 7 3 5.0E-2 5.0E-4 20 0.90 7 4 5.0E-2 5.0E-4 21 0.95 7 5 5.0E-2 5.0E-4 22 0.93 7 6 5.0E-2 5.0E-4 23 0.94 7 7 5.0E-2 5.0E-4 24 0.70 5 8 5.0E-2 5.0E-4 25 0.90 5 9 5.0E-2 5.0E-4 26 0.92 5 10 5.0E-2 5.0E-4 27 0.92 5 11 5.0E-2 5.0E-4 28 0.95 5 12 5.0E-2 5.0E-4 29 0.88 3 13 5.0E-2 5.0E-4 30 0.91 4 14 5.0E-2 5.0E-4 31 0.95 4 15 5.0E-2 5.0E-4 32 0.91 4 Thermal conductivity (W m\(^{-1}\) K\(^{-1}\)):
pavement_type layer 1 layer 2 layer 3 layer 4 layer 5 layer 6 layer 7 layer 8 1 1.00 1.00 1.00 2.10 2.10 0.40 0.40 - 2 0.82 0.82 0.82 2.10 2.10 0.40 0.40 - 3 1.51 1.51 1.51 2.10 2.10 0.40 0.40 - 4 2.19 2.19 2.19 2.19 2.10 0.40 0.40 - 5 2.19 2.19 2.19 2.19 2.10 0.40 0.40 - 6 2.19 2.19 2.19 2.19 2.10 0.40 0.40 - 7 53.3 53.3 53.3 53.3 53.3 - - - 8 0.12 0.12 0.12 0.12 0.12 - - - 9 1.40 1.40 1.40 1.40 1.40 - - - 10 1.40 1.40 1.40 1.40 1.40 - - - 11 1.40 1.40 1.40 1.40 1.40 - - - 12 0.20 0.20 0.20 - - - - - 13 0.12 0.12 0.12 0.12 - - - - 14 0.42 0.42 0.42 0.42 - - - - 15 0.84 0.84 0.84 0.84 - - - - Heat capacity (J K\(^{-1}\) m\(^{-3}\)):
pavement_type layer 1 layer 2 layer 3 layer 4 layer 5 layer 6 layer 7 layer 8 1 2.00E6 2.00E6 2.00E6 2.00E6 2.00E6 1.40E6 1.40E6 - 2 1.94E6 1.94E6 1.94E6 2.00E6 2.00E6 1.40E6 1.40E6 - 3 2.11E6 2.11E6 2.11E6 2.00E6 2.00E6 1.40E6 1.40E6 - 4 2.25E6 2.25E6 2.25E6 2.00E6 2.00E6 1.40E6 1.40E6 - 5 2.25E6 2.25E6 2.25E6 2.00E6 2.00E6 1.40E6 1.40E6 - 6 2.25E6 2.25E6 2.25E6 2.00E6 2.00E6 1.40E6 1.40E6 - 7 3.93E6 3.93E6 3.93E6 3.93E6 3.93E6 - - - 8 0.66E6 0.66E6 0.66E6 0.66E6 0.66E6 - - - 9 1.76E6 1.76E6 1.76E6 1.76E6 1.76E6 - - - 10 1.76E6 1.76E6 1.76E6 1.76E6 1.76E6 - - - 11 1.76E6 1.76E6 1.76E6 1.76E6 1.76E6 - - - 12 0.50E6 0.50E6 0.50E6 - - - - - 13 2.12E6 2.12E6 2.12E6 2.12E6 - - - - 14 0.63E6 0.63E6 0.63E6 0.63E6 - - - - 15 1.77E6 1.77E6 1.77E6 1.77E6 - - - -
residual_moisture#
- Fortran Type: R
- Default: Depends on soil_type
-
SI-Unit: m\(^3\) m\(^{-3}\)
Residual moisture content in the soil model.
root_fraction#
- Fortran Type: R (21)
-
Default: Depends on vegetation_type
Root distribution (\(R\)) in the soil in terms of a root fraction.
A value must be assigned to each soil layer defined by dz_soil. For N soil layers with index k it must hold that \(\sum_{k=1}^{N} R_k =1\). The root fraction it automatically set based on the setting of vegetation_type using the following look-up table for a default four-layer soil configuration using linear interpolation:
vegetation_type Layer 1 Layer 2 Layer 3 Layer 4 (0.00 - 0.07 m) (0.07 - 0.28 m) (0.28 - 1.00 m) (1.00 - 2.89 m) 1 0.00 0.00 0.00 0.00 2 0.24 0.41 0.31 0.04 3 0.35 0.38 0.23 0.04 4 0.26 0.39 0.29 0.06 5 0.26 0.38 0.29 0.07 6 0.24 0.38 0.31 0.07 7 0.25 0.34 0.27 0.14 8 0.27 0.27 0.27 0.09 9 1.00 0.00 0.00 0.00 10 0.47 0.45 0.08 0.00 11 0.24 0.41 0.31 0.04 12 0.17 0.31 0.33 0.19 13 0.00 0.00 0.00 0.00 14 0.25 0.34 0.27 0.11 15 0.23 0.36 0.30 0.11 16 0.23 0.36 0.30 0.11 17 0.19 0.35 0.36 0.10 18 0.19 0.35 0.36 0.10
saturation_moisture#
- Fortran Type: R
- Default: Depends on soil_type
-
SI-Unit: m\(^3\) m\(^{-3}\)
Soil moisture at saturation in the soil model.
lsm_start_time#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: s
Time at which the land surface model is switched on.
This parameter can be used to run PALM for a period of time without the land surface model, e.g. for letting the model develop turbulence before it interacts with the land surface.
soil_moisture#
- Fortran Type: R (21)
- Default: 0.0
-
SI-Unit: m\(^3\) m\(^{-3}\)
Initial soil moisture content distribution.
Volumetric soil moisture content distribution over N soil layers as defined by dz_soil. This parameter is only considered when humidity = .T.. By default, the soil is completely dry. The soil moisture should be set smaller than saturation_moisture. The setting of soil_moisture can be overwritten if respective data is provided by the dynamic driver file.
soil_temperature#
- Fortran Type: R (21)
- Default: undefined
-
SI-Unit: K
Soil temperature distribution.
Soil temperature distribution over N soil layers. The soil temperatures are defined at the center of the layers as defined by dz_soil. The setting of the soil temperature is mandatory unless it is provided by a dynamic driver file.
soil_type#
- Fortran Type: I
-
Default: 3
Soil type used by the soil model according to internal classification.
The user can choose from six soil types according to the ECMWF-IFS classification (parameterization after Van Genuchten, 1980) based on soil porosity. The setting of soil_type can be overwritten if respective data is provided by the static driver file.
soil_type Description Notes 1 Coarse 2 Medium 3 Medium-fine 4 Fine 5 Very fine 6 Organic By setting the soil type, the following parameters will automatically be set:
soil_type alpha_vg l_vg n_vg gamma
_w_satm_sat m_fc m_wilt m_res (m s\(^{-1}\)) (m\(^3\) m\(^{-3}\)) (m\(^3\) m\(^{-3}\)) (m\(^3\) m\(^{-3}\)) (m\(^3\) m\(^{-3}\)) 1 3.83 1.150 1.38 6.94E-6 0.403 0.244 0.059 0.025 2 3.14 -2.342 1.28 1.16E-6 0.439 0.347 0.151 0.010 3 0.83 -0.588 1.25 0.26E-6 0.430 0.383 0.133 0.010 4 3.67 -1.977 1.10 2.87E-6 0.520 0.448 0.279 0.010 5 2.65 2.500 1.10 1.74E-6 0.614 0.541 0.335 0.010 6 1.30 0.400 1.20 1.20E-6 0.766 0.663 0.267 0.010
surface_type#
- Fortran Type: C*10
-
Default: read_from_file
Prescribed surface type in the land surface model based on internal classification.
Prescribed surface type in the land surface model. Allowed values are 'vegetation', 'pavement', 'water', and 'read_from_file' (deprecated setting 'netcdf' is still working but will be removed in the future). Depending on the setting of surface_type, the required parameters are set via vegetation_type, pavement_type, or water_type. If the parameter is set to 'read_from_file', all required parameters are provided from a static driver file (e.g. for prescribing heterogeneous surface configurations).
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Switch off land surface model, despite the Namelist is set.
vegetation_coverage#
- Fortran Type: R
-
Default: Depends on vegetation_type
Vegetation coverage on the surface.
This parameter can have values in the range of (0.0-1.0) and is treated per surface element. The residual fraction is considered to be bare soil. For small grid spacings, the vegetation coverage can usually be considered to be 1 (i.e. the surface element is 100% covered with vegetation).
vegetation_type#
- Fortran Type: I
-
Default: 2
Vegetation type according to internal classification.
Vegetation type according to internal classification used in the land surface model. The user can choose from 19 pre-defined classes similar to the ECMWF-IFS model:
vegetation_type Description Notes 1 bare soil 2 crops, mixed farming 3 short grass 4 evergreen needleleaf trees 5 deciduous needleleaf trees 6 evergreen broadleaf trees 7 deciduous broadleaf trees 8 tall grass 9 desert 10 tundra 11 irrigated crops 12 semidesert 13 ice caps and glaciers Not yet implemented 14 bogs and marshes 15 evergreen shrubs 16 deciduous shrubs 17 mixed forest/woodland 18 interrupted forest By setting vegetation_type, the following parameters are automatically set:
vegetation_type r_c_min LAI c_veg gD z0_vegetation z0h_vegetation lambda_s lambda_u f_sw_in c_surface albedo_type emissivity (s m\(^{-1}\)) (m\(^2\) m\(^{-2}\)) (hPa\(^{-1}\)) (m) (m) (W m\(^{-2}\) K\(^{-1}\)) (W m\(^{-2}\) K\(^{-1}\)) (J m\(^{-2}\) K\(^{-1}\)) 1 0.0 0.00 0.00 0.00 0.005 0.5E-4 0.0 0.0 0.00 0.0 17 0.94 2 180.0 3.00 1.00 0.00 0.1 0.0001 10.0 10.0 0.05 0.0 2 0.95 3 110.0 2.00 1.00 0.00 0.03 0.3E-4 10.0 10.0 0.05 0.0 5 0.95 4 500.0 5.00 1.00 0.03 2.0 2.0 20.0 15.0 0.03 0.0 6 0.97 5 500.0 5.00 1.00 0.03 2.0 2.0 20.0 15.0 0.03 0.0 8 0.97 6 175.0 5.00 1.00 0.03 2.0 2.0 20.0 15.0 0.03 0.0 9 0.97 7 240.0 6.00 0.99 0.13 2.0 2.0 20.0 15.0 0.03 0.0 7 0.97 8 100.0 2.00 0.70 0.00 0.47 0.47E-2 10.0 10.0 0.05 0.0 10 0.97 9 250.0 0.05 0.00 0.00 0.013 0.013E-2 15.0 15.0 0.00 0.0 11 0.94 10 80.0 1.00 0.50 0.00 0.034 0.034E-2 10.0 10.0 0.05 0.0 13 0.97 11 180.0 3.00 1.00 0.00 0.5 0.5E-2 10.0 10.0 0.05 0.0 2 0.97 12 150.0 0.50 0.10 0.00 0.17 0.17E-2 10.0 10.0 0.05 0.0 11 0.97 13* 0.0 0.00 0.00 0.00 1.3E-3 1.3E-4 58.0 58.0 0.00 0.0 14 0.97 14 240.0 4.00 0.60 0.00 0.83 0.83E-2 10.0 10.0 0.05 0.0 3 0.97 15 225.0 3.00 0.50 0.00 0.1 0.1E-2 10.0 10.0 0.05 0.0 4 0.97 16 225.0 1.50 0.50 0.00 0.25 0.25E-2 10.0 10.0 0.05 0.0 5 0.97 17 250.0 5.00 1.00 0.03 2.0 2.0 20.0 15.0 0.03 0.0 10 0.97 18 175.0 2.50 1.00 0.03 1.1 1.1 20.0 15.0 0.03 0.0 7 0.97 *) dummy value for this class has not been implemented in PALM yet.
water_temperature#
- Fortran Type: R
- Default: Depends on water_type
-
SI-Unit: K
Initial water temperature.
Initial water body temperature, as prescribed by water_type. Note that water bodies are not interactive in PALM at the moment so that the water temperature remains constant during the simulation unless the user prescribes temporal changes in the water body temperature via the user interface.
water_type#
- Fortran Type: I
-
Default: 1
Water body type based on internal classification.
Water body type based on PALM's classification. By setting water_type, the following parameters are automatically set:
water_type Description water_temperature z0_water z0h_water lambda_s lambda_u albedo_type emissivity (K) (m) (m) (W m\(^{-2}\) K\(^{-1}\)) (W m\(^{-2}\) K\(^{-1}\)) 1 Lake 283.0 0.001 0.00001 1E10 1E10 1 0.95 2 River 283.0 0.003 0.00003 1E10 1E10 1 0.95 3 Ocean 283.0 0.001 0.00001 1E10 1E10 1 0.95 4 Pond 283.0 0.001 0.00001 1E10 1E10 1 0.95 5 Fountain 283.0 0.01 0.001 1E10 1E10 1 0.95
wilting_point#
- Fortran Type: R
- Default: Depends on soil_type
-
SI-Unit: m\(^3\) m\(^{-3}\)
Volumetric soil moisture content at permanent wilting point as used in the soil model.
z0_pavement#
- Fortran Type: R
- Default: Depends on pavement_type
-
SI-Unit: m
Roughness length for momentum for pavements.
Roughness length for momentum for a surface classified as pavement via the setting of surface_type = 'pavement'. The setting of this parameter overwrites the default setting via pavement_type.
z0_vegetation#
- Fortran Type: R
- Default: Depends on vegetation_type
-
SI-Unit: m
Roughness length for momentum for vegetation.
Roughness length for momentum for a surface classified as vegetation via the setting of surface_type = 'vegetation'. The setting of this parameter overwrites the default setting via vegetation_type.
z0_water#
- Fortran Type: R
- Default: Depends on water_type
-
SI-Unit: m
Roughness length for momentum for water.
Roughness length for momentum for a surface classified as water via the setting of surface_type = 'water'. The setting of this parameter overwrites the default setting via water_type.
z0h_pavement#
- Fortran Type: R
- Default: Depends on pavement_type
-
SI-Unit: m
Roughness length for heat for pavements.
Roughness length for heat for a surface classified as pavement via the setting of surface_type = 'pavement'. The setting of this parameter overwrites the default setting via pavement_type.
z0h_vegetation#
- Fortran Type: R
- Default: Depends on vegetation_type
-
SI-Unit: m
Roughness length for heat for vegetation.
Roughness length for heat for a surface classified as vegetation via the setting of surface_type = 'vegetation'. The setting of this parameter overwrites the default setting via vegetation_type.
z0h_water#
- Fortran Type: R
- Default: Depends on water_type
-
SI-Unit: m
Roughness length for heat for waters.
Roughness length for heat for a surface classified as water via the setting of surface_type = 'water'. The setting of this parameter overwrites the default setting via water_type.
z0q_pavement#
- Fortran Type: R
- Default: Value of z0h_pavement
-
SI-Unit: m
Roughness length for moisture for pavements.
Roughness length for moisture for a surface classified as pavement via the setting of surface_type = 'pavement'. The setting of this parameter overwrites the default setting via pavement_type. By default, z0q_pavement is set equal to the value of z0h_pavement.
z0q_vegetation#
- Fortran Type: R
- Default: Value of z0h_vegetation
-
SI-Unit: m
Roughness length for moisture for pavements.
Roughness length for moisture for a surface classified as vegetation via the setting of surface_type = 'vegetation'. The setting of this parameter overwrites the default setting via vegetation_type. By default, z0q_vegetation is set equal to the value of z0h_vegetation.
z0q_water#
- Fortran Type: R
- Default: Value of z0h_water
-
SI-Unit: m
Roughness length for moisture for water.
Roughness length for moisture for a surface classified as water via the setting of surface_type = 'water'. The setting of this parameter overwrites the default setting via water_type. By default, z0q_water is set equal to the value of z0h_water.
Offline Nesting Parameters#
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Switch to turn off the module.
If set to .T., mesoscale nesting is not activated even if the namelist is present in the parameter file.
Nesting Parameters#
anterpolation_buffer_width#
- Fortran Type: I
-
Default: 2
Width of the anterpolation boundary buffer in number of parent-grid points.
If anterpolation_buffer_width = 0, anterpolation is carried out for all parent-grid points covered by a child domain except the grid points next to the boundaries. Setting anterpolation_buffer_width > 0 widens these buffers to the specified value. The same value is applied to all nested boundaries. Enlarging the buffer may help to prevent unwanted feedbacks between parent and child in case of nesting_mode = 'two-way'.
Note:
In ocean mode, anterpolation_buffer_width = 0 may cause aborts due to exponentially increasing velocities.
anterpolation_starting_height#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: m
Lower level at which anterpolation is employed.
This parameter controls the canopy-restricted anterpolation. It describes the height at which the anterpolation is performed in case of two-way coupling mode. Below this height, no anterpolation is employed. It is recommended to set anterpolation_starting_height to a height lower than the majority of the building roofs and terrain features. Note that in vertically shifted childs anterpolation_starting_height still refers to the coordinate system of the root domain.
domain_layouts#
- Fortran Type: D (64)
-
Default: undefined
General information to describe the nesting setup.
The derived data type has the following structure:
TYPE pmc_layout CHARACTER(LEN=32) :: name INTEGER :: id INTEGER :: parent_id INTEGER :: npe_total REAL(wp) :: lower_left_x REAL(wp) :: lower_left_y REAL(wp) :: nest_shift_z END TYPE pmc_layoutnameis the domain name given by the user.iddescribes the number of the domain (this can be arbitrarily chosen, but it is good practice to number the domains in ascending order starting with 1), whileparent_idis the number of the domain in which the nested domain is embedded. The so-called root domain, which includes all other nested-domains, should have theid = 1. The root domain has no parent domain. Theparent_idof the root domain must be set to -1.npe_totalis the number of PEs to be assigned for this domain.lower_left_xandlower_left_yare the coordinates (in meters) of the south-left corner of the domain in the root-model coordinate system. Note that the corner coordinates are always given in the root-coordinate system even for domains nested in another nested domain. More precisely,lower_left_xis measured from the u-node ati = 0andlower_left_yfrom the v-node atj = 0. The root-domain haslower_left_x = 0.0andlower_left_y = 0.0. Note thatlower_left_xmust be integer divisible by the parent grid dx and, similarly,lower_left_yby the parent grid dy.nest_shift_zdescribes the vertical shift (in meters) of a child with respect to the root domain.nest_shift_z = 0.0means that the bottom boundary of the child matches the bottom boundary of the root domain. The root domain always requires to setnest_shift_z = 0.0. In ocean mode,nest_shift_z /= 0.0is not allowed, and the top boundary of the child always matches the top boundary of the root model (the atmosphere/ocean surface). The bottom coordinate of a child (as given bynest_shift_z) must match a vertical grid level of the root domain. If e.g. the vertical grid spacing of the root domain is dz = 20.0, only values of 20.0, 40.0, 60.0, etc. are allowed fornest_shift_z.A nested domain must be completely embedded within its parent domain and there must be a clearance between the lateral nest boundaries and its parent boundaries. This clearance must be at least four parent-grid cells if using the Wicker-Skamarock advection scheme and at least two parent-grid cells if using the Piaseck-Williams advection scheme. Possible parallel nests, and the associated ghost-cell layers, are not allowed to overlap.
Up to 64 domains can be used.
The following gives an example for defining two nested-domains embedded in a root-domain, where the second child (
'fine') is embedded in the first child ('medium'):domain_layouts = 'coarse', 1, -1, 16, 0.0, 0.0, 0.0, 'medium', 2, 1, 16, 320.0, 128.0, 0.0, 'fine', 3, 2, 16, 480.0, 160.0, 100.0,The total number of MPI-processes required by this configuration is 16 + 16 + 16 = 48. The domain decomposition along x- and y-directions of each domain can be additionally specified by setting npex and npey in the respective runtime-parameters namelists.
homogeneous_initialization_child#
- Fortran Type: L
-
Default: .FALSE.
Child-domain initialization method.
In case of homogeneous_initialization_child = .F., the child domain is initialized by the 3D data received from its parent domain.
If large differences between parent and child domains appear in the topography, grid points might belong to the atmosphere in the child domain but the corresponding parent grid points belong to topography. With a 3D initialization, topography-covered parent grid points will then be visible in the child domain as regions with zero values of the velocity components. In case the discrepancy between parent and child topography becomes excessively large, i.e. for larger grid-aspect ratios, this causes issues with the mass balance, which in turn may result in numerical instabilities.
To overcome this, the nesting offers the possibility to initialize the child domain with area-averaged parent data (homogeneous_initialization_child = .T.), with area averaging performed over the respective child area.
homogeneous_initialization_child = .T. is not allowed in ocean mode.
nesting_datatransfer_mode#
- Fortran Type: C*7
-
Default: mixed
Type of nesting data-transfer mode.
This parameter is only relevant for recursively nested domains where at least one child domain is also the parent domain for another child domain and nesting_mode = 'two-way'.
Currently 3 choices are available:
-
cascade
From coarse to fine: The child waits until it has received data from the coarse model, performs the interpolation, and then sends data to the finer model.
From fine to coarse: The parent waits until it has received anterpolated data from the finer model, performs further anterpolation, and then sends the data to the coarser grid.
-
mixed
From coarse to fine: The child receives data from the coarse model and performs the interpolation, while it sends data to its recursively nested child domain without waiting until interpolation has been finished.
From fine to coarse: The parent receives anterpolated data from its child, while it sends data data to the coarser domain simultaneously.
-
overlap
From coarse to fine: All parent domains immediately send data after timestep synchronization. The respective child domain receives the data via 'MPI_GET' and perform the interpolation.
From fine to coarse: The anterpolation step is also performed simultaneously for all models. Subsequently, the data is transferred to the coarse model in parallel.
-
nesting_bounds#
- Fortran Type: C*14
-
Default: 3d_nested
Type of nesting boundary conditions.
Currently 4 choices are available:
-
3d_nested
A full 3d-nested simulation. Dirichlet boundary conditions are employed at the east, west, north, south and top domain child boundaries.
-
cyclic_along_x
The boundary condition at the east and west domain boundaries is cyclic, while Dirichlet boundary conditions are employed at the north, south and top domain boundaries. The child domain extension must exactly match the root domain extension along the
xdirection. -
cyclic_along_y
The boundary condition at the north and south domain boundaries is cyclic, while Dirichlet boundary conditions are employed at the east, west and top domain boundaries. The child domain extension must exactly match the root domain extension along the
ydirection. -
vertical_only
A one-dimensional nesting in the vertical direction only, i.e. all domains (must) have the same horizontal extent.
-
nesting_mode#
- Fortran Type: C*7
-
Default: two-way
Type of nesting mode.
Currently 2 choices are available:
-
one-way
A one-way nesting is applied. The child domain obtains its boundary conditions from the parent domain via interpolation but there is no feedback of the child-domain solution on the parent.
-
two-way
A two-way nesting is applied. The child domain obtains its boundary conditions from the parent domain via interpolation and feeds back its solution onto the parent domain via anterpolation.
-
particle_coupling#
- Fortran Type: L
-
Default: .TRUE.
Switch for particle coupling in nested setups.
This parameter takes effect only when the Lagrangian particle model is activated. For particle_coupling = .T., Lagrangian particles are transferred between domains. Particles only exist once, either in the parent or in the child domain, i.e. if a particle enters a child domain, it is removed from the parent, and if it leaves the child, it is removed from the child and added to the parent domain again. The &particle_parameters namelist is only allowed to be given for the root domain.
For particle_coupling = .F., &particle_parameters namelists can be given independently for each domain. Particles will not be transferred between parent and child domains. Boundary conditions for particles are assumed as set in the respective &particle_parameters namelist.
particle_coupling = .T. is not allowed in case that vertically shifted child domains are used.
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Switch to turn off the module.
If switch_off_module = .T., the nesting is not activated even if the namelist is present in the parameter file.
Ocean Parameters#
bc_sa_t#
- Fortran Type: C*20
-
Default: neumann
Top boundary condition of the salinity.
When a constant salinity flux is used at the top boundary (see top_salinityflux), bc_sa_t = 'neumann' must be used, because otherwise the resolved scale may contribute to the top flux so that a constant value of the flux cannot be guaranteed.
Currently 2 choices are available:
-
dirichlet
Salinity at the top (
sa(k=nz+1), i.e. sea surface) does not change during the run. -
neumann
Vertical gradient of the salinity at the top is zero (
sa(k=nz+1) = sa(k=nz)).
-
bottom_salinityflux#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: psu ms-1
Kinematic salinity flux near the ocean bottom.
The respective salinity flux value is used as the bottom (horizontally homogeneous) boundary condition for the salinity equation. This additionally requires that a Neumann condition must be used for the salinity, which is currently the only available bottom boundary condition (so it does not need to be set).
See also top_salinityflux.
salinity#
- Fortran Type: L
-
Default: .TRUE.
Parameter to switch on/off calculation of the salinity equation.
You may set salinity = .F. in ocean mixed layer studies (where the turbulent layer is shallow) to save CPU time. In that case, a constant salinity as given by sa_surface is assumed when calculating the equation of state.
In the case of salinity = .F., non-zero values for parameters bottom_salinityflux and top_salinityflux are not allowed.
sa_surface#
- Fortran Type: R
- Default: 35.0
-
SI-Unit: psu
Ocean surface salinity.
This parameter is used to set the value of the salinity
saat the sea surface (vertical grid index k=nzt). Starting from this value, the initial vertical salinity profile is constructed from the sea surface down to the bottom of the sea (vertical grid indexk=0) by using sa_vertical_gradient and sa_vertical_gradient_level.
sa_vertical_gradient#
- Fortran Type: R (10)
- Default: 0.0
-
SI-Unit: psu / 100 m
Salinity gradient(s) of the initial salinity profile.
This salinity gradient holds starting from the height level defined by sa_vertical_gradient_level down to the bottom boundary or down to the next height level defined by sa_vertical_gradient_level. More precisely, for all uv levels, k, where zu(k) < sa_vertical_gradient_level, sa_init(k) is set: sa_init(k) = sa_init(k+1) - dzu(k+1) * sa_vertical_gradient. A total of 10 different gradients for 11 height intervals (10 intervals if sa_vertical_gradient_level(1) = 0.0) can be assigned. The surface salinity at k=nzt is assigned via sa_surface.
Example:
> sa_vertical_gradient = *1.0*, *0.5*, > sa_vertical_gradient_level = *-500.0*, *-1000.0*,The example defines the salinity to be constant down to z = -500.0 m with a salinity given by sa_surface. For -500.0 m < z <= -1000.0 m the salinity gradient is 1.0 psu / 100 m and for z < -1000.0 m down to the bottom boundary it is 0.5 psu / 100 m (it is assumed that the assigned height levels correspond with uv levels).
sa_vertical_gradient_level#
- Fortran Type: R (10)
- Default: 0.0
-
SI-Unit: m
Height level from which on the salinity gradient defined by sa_vertical_gradient is effective.
The height levels must be assigned in descending order. The default values result in a constant salinity profile regardless of the values of sa_vertical_gradient (unless the bottom boundary of the model is lower than -100000.0 m). For the piecewise construction of salinity profiles see sa_vertical_gradient.
stokes_waveheight#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: m
Parameter to switch on and set the effects of Langmuir circulation in the ocean mode (height of ocean waves).
The effects of Langmuir circulation are considered by the Craik-Leibovich vortex force and the Stokes drift velocity, as described in Noh et al. (2004), JPO. In order to switch these effects on, you need to set both parameters, stokes_waveheight and stokes_wavelength to a non-zero positive value. stokes_waveheight sets the height of the ocean surface waves to be used for calculating the vertical profile of the Stokes drift velocity. The direction of the Stokes drift velocity is derived from parameters top_momentumflux_u and top_momentumflux_v, which define the wind stress at the ocean surface.
stokes_wavelength#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: m
Parameter to switch on and set the effects of Langmuir circulation in the ocean mode (wavelength of ocean waves).
stokes_wavelength sets the wavelength of the ocean surface waves to be used for calculating the vertical profile of the Stokes drift velocity. See stokes_waveheight for more information about how the Stokes drift is determined.
surface_cooling_spinup_time#
- Fortran Type: R
- Default: undefined
-
SI-Unit: s
Time after which the surface heat flux is switched off.
Limits the duration of a surface heat flux forcing. If, for example, a surface cooling is required during an initial spin-up phase for only 15 minutes in order to trigger the development of a Langmuir circulation, the heat flux must be given via top_heatflux and surface_cooling_spinup_time = 900.0 must be set. The default value means that the surface flux is (probably) never switched off.
Attention:
In nested runs surface_cooling_spinup_time should be given the same value in all domains (root domain as well as child domains).
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Parameter to switch on/off the module in general.
switch_off_module = .T. can be used to switch off this module without removing (or commenting) the namelist from the namelist file.
top_salinityflux#
- Fortran Type: R
- Default: undefined
-
SI-Unit: psu ms-s
Kinematic salinity flux at the top boundary, i.e. the sea surface.
The respective salinity flux value is used as the top (horizontally homogeneous) boundary condition for the salinity equation. This additionally requires that a Neumann condition for the salinity (see bc_sa_t has been set, because otherwise the resolved scale transport may contribute to the top flux so that a constant salinity flux cannot be guaranteed.
The application of a salinity flux at the model top additionally requires the setting of the initial parameter use_top_fluxes = .T.
See also bottom_salinityflux.
wave_breaking#
- Fortran Type: L
-
Default: .FALSE.
Switches on turbulence generation by wave breaking.
wave_breaking = .T. switches on turbulence generation by wave breaking at the ocean surface. This way, random perturbations are added to the horizontal velocity components at the top model layer (at grid index k = nzt). The parameterization is realized as described in Noh et al. (2004), JPO.
WARNING: The implemented parameterization is designed for a vertical grid spacing of dz = 1.25 m and time steps of about 4 s. It will probably fail for other setups.
Particle Parameters#
aero_species#
- Fortran Type: C*15
-
Default: nacl
Define chemical composition of aerosol particles.
Depending on the chosen chemical composition, the van't Hoff factor, weight of solute and the density of the aerosol particles are defined.
This parameter comes only into play if curvature_solution_effects = .T. and the particle model is used as cloud model, i.e. cloud_droplets = .T..
Currently 3 choices are available:
-
nacl
Sodium Chloride
-
c3h4o4
Malonic Acid
-
nh4o3
Ammonium Ozonide
-
aero_type#
- Fortran Type: C*15
-
Default: maritime
Type of aerosols to be considered.
Depending on the type, the aerosol spectrum is constructed using three log-normal distributions and the values stated in Jaenicke (1993).
The aerosol spectrum determines the aerosol mass, initial wet radius, and the weighting factor of all super-droplets. The weighting factors can be manipulated with aero_weight.
Note that curvature_solution_effects needs to be true to consider aerosol spectra and the particle model must be used as cloud model, i.e. cloud_droplets = .T..
Currently 8 choices are available:
-
background
Typical background profiles
-
continental
Typical continental profiles
-
desert
Typical desert profiles
-
maritime
Typical maritime profiles
-
polar
Typical polar profiles
-
rural
Typical rural profiles
-
urban
Typical urban profiles
-
user
If 'user' is chosen, the aerosol spectrum is steered by rm, na, and log_sigma.
-
aero_weight#
- Fortran Type: R
-
Default: 1.0
Scaling parameter for the aerosol number concentration.
Scale the number concentration defined by aero_type to the desired amount of aerosols by keeping the typical shape of the spectrum.
alloc_factor#
- Fortran Type: R
- Default: 20.0
-
SI-Unit: %
Factor describing the memory allocated additionally to the memory needed for initial particles at a given grid cell.
If 100 particles are initialized at a grid cell, then an array for 120 particles is allocated (if alloc_factor = 20). This provides sufficient memory for particles being transported to this grid cell during the simulation.
Note that the particle array is automatically reallocated automatically if more particles are transported to this array than its current size. Thus, it is not necessary to choose alloc_factor too high.
bc_par_b#
- Fortran Type: C*15
-
Default: reflect
Bottom boundary condition for particle transport.
Currently 3 choices are available:
-
absorb
The particles are absorbed at the bottom boundary.
-
reflect
The particles are reflected at the bottom boundary.
-
reset
The particles are randomly reset within the particle layer. This method might be appropriate in case of simulating cloud droplets and mitigate superdroplet (and therefore aerosol) removal by sedimentation and precipitation.
-
bc_par_lr#
- Fortran Type: C*15
-
Default: Depends on bc_lr
Lateral boundary condition (x-direction) for particle transport.
This lateral boundary condition should correspond to the lateral boundary condition used for the flow (see bc_lr).
In case of a nested run, the default value of bc_par_lr is 'nested' in the nest domains. For the root domain, the default is 'cyclic'.
Currently 4 choices are available:
-
absorb
The particles are absorbed at the left and right boundary.
-
cyclic
In non-nested setups, this is the default value. Cyclic boundary conditions are applied at the left and right boundary.
-
nested
Particles are transferred to the parent domain at the left and right boundary.
-
reflect
The particles are reflected at the left and right boundary.
-
bc_par_ns#
- Fortran Type: C*15
-
Default: Depends on bc_ns
Lateral boundary condition (y-direction) for particle transport.
This lateral boundary condition should correspond to the lateral boundary condition used for the flow (see bc_ns).
In case of a nested run, the default value of bc_par_ns is 'nested' in the nest domains. For the root domain, the default is 'cyclic'.
Currently 4 choices are available:
-
absorb
The particles are absorbed at the North and South boundary.
-
cyclic
In non-nested setups, this is the default value. Cyclic boundary conditions are applied at the North and South boundary.
-
nested
Particles are transferred to the parent domain at the North and South boundary.
-
reflect
The particles are reflected at the North and South boundary.
-
bc_par_t#
- Fortran Type: C*15
-
Default: absorb
Top boundary condition for particle transport.
In case of a nested run, the default value is 'nested' in the nest domains. For the root domain, the default is 'absorb'.
Currently 2 choices are available:
-
absorb
The particles are absorbed at the top boundary.
-
reflect
The particles are reflected at the top boundary.
-
collision_kernel#
- Fortran Type: C*15
-
Default: none
Parameter to steer cloud droplet growth by collision processes.
The growth of cloud droplets due to collision is parameterized using the so-called collision kernel. This parameter is only effective if cloud_droplets = .T.. Attention: Simulating the collision process drastically increases the CPU time.
Currently 5 choices are available:
-
hall
Collision kernel from Hall (1980), which considers collision due to pure gravitational effects. Larger droplets have a higher terminal fall velocity and are collecting smaller ones. Only terminal droplet velocities are considered in this kernel, not their effective velocities.
-
hall_fast
Same as 'hall', but a collision efficiency table is calculated at the start of the simulation for fixed radius classes in the range of 10-6 m and 10-3 m. The number of classes to be used (i.e. the resolution of the kernel) can be set by parameter radius_classes. This method requires significantly less CPU time than the 'hall' method.
-
none
Droplet collision is switched off.
-
wang
Beside gravitational effects (treated with the Hall kernel) also the effects of turbulence on the collision are considered using parameterizations of Ayala et al. (2008) and Wang and Grabowski (2009).
This kernel includes three possible effects of turbulence:
- the modification of the relative velocity between the droplets,
- the effect of preferential concentration, and
- the enhancement of collision efficiencies.
-
wang_fast
Same as 'wang' , but a collision efficiency table is calculated at the start of the simulation for fixed radius- and dissipation classes in the of 10-6 m and 10-3 m, and 0 and 600 cm2 s-3, respectively. The number of classes to be used (i.e. the resolution of the kernel) can be set by parameters radius_classes, and dissipation_classes. This method requires significantly less CPU time than the 'wang' method.
-
curvature_solution_effects#
- Fortran Type: L
-
Default: .FALSE.
Parameter to consider solution and curvature effects on the equilibrium vapor pressure of cloud droplets.
If switched on, the radius growth equation is integrated in time using a Rosenbrock method. This parameter is only effective if cloud_droplets = .T..
data_output_pts#
- Fortran Type: C*30 (50)
-
Default: ''
List of particle-output quantities.
deallocate_memory#
- Fortran Type: L
-
Default: .TRUE.
Parameter to enable deallocation of unused memory.
If the number of particles in a grid box exceeds the allocated memory, new memory is allocated. However, in case the number of particles per grid box is only temporarily high, most of the memory remains unused.
If deallocate_memory = .T., the allocated memory used for particles will be dynamically adjusted with respect to the current number of particles after a given number of timesetps (see step_dealloc).
density_ratio#
- Fortran Type: R (10)
-
Default: 0.0
Ratio of the density of the fluid and the density of the particles.
If density_ratio = 0.0, particles are weightless and are transported passively with the resolved scale flow.
If density_ratio \(\neq\) 0.0, particles have a mass and hence inertia so that their velocity deviates from the velocity of the surrounding flow. Particle velocity is calculated analytically. In this case, the particle radius as well as the molecular viscosity (1.461E-5 m2 s-1) also affect the velocity.
If density_ratio = 1.0, the particle density corresponds to the density of the surrounding fluid and the particles do not feel any buoyancy.
If density_ratio > 1.0, particles will be accelerated upwards.
If density_ratio < 1.0, particles will be accelerated downwards.
With several groups of particles (see number_of_particle_groups), each group can be assigned a different value. If the number of values given for density_ratio is less than the number of groups defined by number_of_particle_groups), then the last assigned value is used for all remaining groups.
dissipation_classes#
- Fortran Type: I
-
Default: 10
Number of dissipation classes to be used in the collision-efficiency table.
This parameter comes into effect if a fast method is used for the collision_kernel (flagged as '_fast'). It defines the number of dissipation classes which spawn the collision-efficiency table. The interval [1.0, 1000.0] cm s-3 is divided into n = dissipation_classes equidistant parts.
dt_dopts#
- Fortran Type: R
- Default: Value of dt_data_output
-
SI-Unit: s
Temporal interval at which time series data of particle quantities shall be output.
If particle advection is switched on (see dt_prel) this parameter can be used to assign the temporal interval at which time series of particle quantities shall be output. Output is written to DATA_1D_PTS_NETCDF. The file contains time series of statistical quantities and additional optional time series of individual particles, which can be chosen via parameter data_output_pts.
If using multiple particle groups (see number_of_particle_groups), separate time series are output for each group. The particle group is given in the 'long_name' attribute of the netCDF output file which is appended by the string 'PG ##', where '##' is the number of the particle group.
The following list gives a short description of statistical quantities that are always output. Most quantities are averaged over the total number of particles at the specific output time.
netCDF Variable Name Unit Explanation tnpt - total number of particles x_ m particle x-coordinate with respect to the particle origin y_ m particle y-coordinate with respect to the particle origin z_ m particle z-coordinate with respect to the particle origin z_abs m absolute particle z-coordinate u m s-1 u particle velocity component v m s-1 v particle velocity component w m s-1 w particle velocity component u'' m s-1 subgrid-scale u particle velocity component v'' m s-1 subgrid-scale v particle velocity component w'' m s-1 subgrid-scale w particle velocity component npt_up - total number of upward moving particles w_up m s-1 vertical velocity of the upward moving particles w_down m s-1 vertical velocity of the downward moving particles radius_ m particle radius r_min m minimum particle radius r_max m maximum particle radius x*2 m2 variance of the particle x-coordinate with respect to x_ y*2 m2 variance of the particle y-coordinate with respect to y_ z*2 m2 variance of the particle z-coordinate with respect to z_ u*2 m2 s-2 variance of the u particle velocity component with respect to u v*2 m2 s-2 variance of the v particle velocity component with respect to v w*2 m2 s-2 variance of the w particle velocity component with respect to w u"2 m2 s-2 variance of the subgrid-scale u particle velocity component with respect to u" v"2 m2 s-2 variance of the subgrid-scale v particle velocity component with respect to v" w"2 m2 s-2 variance of the subgrid-scale w particle velocity component with respect to w"
dt_min_part#
- Fortran Type: R
- Default: 0.0002
-
SI-Unit: s
Minimum value for the particle time step when subgrid-scale velocities are used.
For details, see use_sgs_for_particles.
dt_prel#
- Fortran Type: R
- Default: 9999999.9
-
SI-Unit: s
Temporal interval at which particles are to be released from a particle source.
Actual release times may slightly deviate from the given time interval (see e.g. dt_dopr).
dt_write_particle_data#
- Fortran Type: R
- Default: 9999999.9
-
SI-Unit: s
Temporal interval for output of particle data.
This parameter can be used to assign the temporal interval at which particle data shall be output. Data are output to the local file PARTICLE_DATA. See the file description for more details about its format.
end_time_prel#
- Fortran Type: R
- Default: 9999999.9
-
SI-Unit: s
Time of the last release of particles.
See also particle_advection_start.
extend_prts_filesize#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: %
Percentage to which the size of the file containing the particle time series shall be enlarged.
By default, the file size for particle time series is determined by the number of output particles that are identified at the beginning of a run (see parameters pts_increment, pts_percentage, and description of input file PARTICLE_IDS).
Since it is not allowed to increase the file size during a run, setting of extend_prts_filesize is required, if the number of output particles increase during a run due to the release of new particle sets. For example, extend_prts_filesize = 50.0 will increase the file size by 50%, so that particles which are newly released at a later time can be output to the file. See also maximum_number_of_output_particles.
It is not allowed to set maximum_number_of_output_particles and extend_prts_filesize at the same time.
initial_weighting_factor#
- Fortran Type: R
- Default: 1.0
-
SI-Unit: 1
Factor to define the real number of initial droplets in a grid box.
In case of explicitly simulating cloud droplets (see cloud_droplets), the real number of initial droplets in a grid box is equal to the initial_weighting_factor times the number of particles in this box defined by the particle-source parameters pdx, pdy, pdz, psb, psl, psr, psn, pss, and pst.
log_sigma#
- Fortran Type: R (3)
- Default: Depends on aero_type
-
SI-Unit: 1
Logarithm (log10) of geometric standard deviation of the log-normal distribution steering the initial dry aerosol spectrum.
See aero_type for more details. Up to three values of log_sigma can be prescribed.
maximum_number_of_output_particles#
- Fortran Type: I
-
Default: 0
Defines the maximum number of particles allowed for output of time series.
maximum_number_of_output_particles must be set, if the number of output particles increase during a run due to the release of new particle sets. It defines the size of the dimension in the netCDF output file.
It is not allowed to increase the size during a run.
It is not allowed to set maximum_number_of_output_particles and extend_prts_filesize at the same time.
By default, the file size is determined by the number of output particles that are identified at the beginning of a run. See also parameters pts_increment, pts_percentage, and description of input file PARTICLE_IDS.
max_number_particles_per_gridbox#
- Fortran Type: I
-
Default: 100
Threshold for splitting of superdroplets.
If the number of particles in a grid box reach max_number_particles_per_gridbox, no more splitting operations (see splitting) are conducted.
Note: This threshold must be given an appropriate value if splitting is switched on. Otherwise new particles will be created in single grid boxes without a limit which may significantly increase the CPU time.
merging#
- Fortran Type: L
-
Default: .FALSE.
Switch on/off merging algorithm for cloud droplets.
This parameter is only effective if cloud_droplets = .T..
The merging algorithm merges two superdroplets into one. It is executed at every timestep. The behaviour can be steered with the parameters radius_merge and weight_factor_merge.
Merging of cloud droplets reduces the amount of computed droplets and hence the demand of computational resources.
Note: Right now, the merging algorithm is only tested for evaporated superdroplets (radius < 0.1 µm) with a low weighting factor (< 0.5 * initial_weighting_factor). For this configuration, micro- and macro-physical quantities of the cloud are not affected by the merging.
na#
- Fortran Type: R (3)
- Default: Depends on aero_type
-
SI-Unit: m-3
Number concentration of the log-normal distribution steering the initial dry aerosol spectrum.
Up to three values of na can be prescribed. See aero_type for more details.
number_concentration#
- Fortran Type: R
- Default: undefined
-
SI-Unit: m-3
Initial particle number concentration.
If this value is set, the initial_weighting_factor is calculated so that the prescribed initial number concentration is ensured in every grid box that contains particles.
number_of_particle_groups#
- Fortran Type: I
-
Default: 1
Number of particle groups to be used.
The maximum allowed number of particle groups is limited to 10.
number_particles_per_gridbox#
- Fortran Type: I
-
Default: undefined
Number of particles which are created in every grid box.
If number_particles_per_gridbox >= 1, the given number of particles are evenly distributed within a grid box if the grid box belongs to the particle-source area (defined by psb, psl, psr, psn, pss, and pst).
If not set, the number of particles per grid box depends on pdx, pdy and pdz.
particles_per_point#
- Fortran Type: I
-
Default: 1
Number of particles to be released per point of the particle source.
particle_advection_interpolation#
- Fortran Type: C*25
-
Default: trilinear
Method for interpolating the flow velocities to the particle position.
Currently 3 choices are available:
-
trilinear
In the trilinear method, the velocities of the closest surrounding grid points are used for interpolation. Particles within a grid box are sorted into eight subgrid boxes to decide which grid points are the closest. This allows for a faster computation of the particle velocity. For more details, see the LPM documentation.
Despite sophisiticated tests and validation, in some cases, this method violates the well-mixed condition.
-
simple_corrector
The simple_corrector considers only the two closest velocity values per direction, i.e. for the u-component the values of
iandi+1are used (for v-component:jandj+1, for w-component:kandk-1).A predictor-corrector method is used, i.e. the interpolation method is done with the values of timestep
tand corrected by the values of timestept+1.For more details, see Grabowski et al., 2018.
-
simple_predictor
The simple_predictor method is the same as the simple_corrector method but without the corrector step.
-
particle_advection_start#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: s
Time of the first release of particles.
By default, particles are released from the beginning of a simulation. If the start of the particle release is set to be after a restart, read_particles_from_restartfile = .F. must be set in the initial run.
See also end_time_prel.
particle_maximum_age#
- Fortran Type: R
- Default: 9999999.9
-
SI-Unit: s
Maximum allowed age of particles.
If the age of a particle exceeds particle_maximum_age, the particle is deleted.
pdx#
- Fortran Type: R (10)
- Default: Value of dx
-
SI-Unit: m
Distance along x between particles within a particle source.
pdx can be assigned a different value for each particle group (see number_of_particle_groups).
pdx is ignored if number_particles_per_gridbox is given.
pdy#
- Fortran Type: R (10)
- Default: Value of dy
-
SI-Unit: m
Distance along y between particles within a particle source.
pdy can be assigned a different value for each particle group (see number_of_particle_groups).
pdy is ignored if number_particles_per_gridbox is given.
pdz#
- Fortran Type: R (10)
- Default: Depends on dz
-
SI-Unit: m
Distance along z between particles within a particle source.
The default is defined as the vertical distance between the two lowest grid levels:
zu(2) - zu(1).pdz can be assigned a different value for each particle group (see number_of_particle_groups).
pdz is ignored if number_particles_per_gridbox is given.
psb#
- Fortran Type: R (10)
- Default: Depends on dz
-
SI-Unit: m
Bottom edge of a particle source.
By default, psb is set to half the domain size:
zu(nz/2). If the local particle source is below the surface, no particle are released at this location.If seed_follows_topography = .T., psb is interpreted as relative to the given topography.
psb can be assigned a different value for each particle group (see number_of_particle_groups).
psl#
- Fortran Type: R (10)
- Default: 0.0
-
SI-Unit: m
Left edge of a particle source.
psl can be assigned a different value for each particle group (see number_of_particle_groups).
psn#
- Fortran Type: R (10)
- Default: Depends on ny
-
SI-Unit: m
Rear ("north") edge of a particle source.
By default, the northern edge of a particle source is equal to the northern domain boundary:
(ny+1)*dy.psn can be assigned a different value for each particle group (see number_of_particle_groups).
psr#
- Fortran Type: R (10)
- Default: Depends on nx
-
SI-Unit: m
Right edge of a particle source.
By default, the right edge of a particle source is equal to the right domain boundary:
(nx+1)*dx.psr can be assigned a different value for each particle group (see number_of_particle_groups).
pss#
- Fortran Type: R (10)
- Default: 0.0
-
SI-Unit: m
Front ("south") edge of a particle source.
pss can be assigned a different value for each particle group (see number_of_particle_groups).
pst#
- Fortran Type: R (10)
- Default: Value of psb
-
SI-Unit: m
Top edge of a particle source.
If the local particle source is below the surface, no particle are released at this location. If seed_follows_topography = .T., pst is interpreted as relative to the given topography.
pst can be assigned a different value for each particle group (see number_of_particle_groups).
pts_increment#
- Fortran Type: I
-
Default: 0
Parameter for steering the number of time-series output of individual particles.
Time series data of every pts_increment-th particle is written to the output file.
By default, no time series is output. If pts_increment = 1, time series of all particles are output; if pts_increment = 2, time series of every second particle is output; etc.
Note: Low values of pts_increment may create huge output and slow down the model performance significantly.
Alternatively, pts_percentage can be used to steer the output of particle time series.
It is not allowed to set both pts_increment and pts_percentage at the same time.
Due to the MPI message size limit of \(2^{31}-1\) elements the number of time-series must not exceed that value.
pts_percentage#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: %
Parameter for steering the number of time-series output of individual particles.
Time series data of the given percentage of the particles is written to the output file.
By default, no time series is output. If pts_percentage = 100, time series data for each particle is output; if pts_percentage = 50, time series of half of all particles is output, etc.
Note: High values of pts_percentage may create huge output and slow down the model performance significantly.
Alternatively, pts_increment can be used to steer the output of particle time series.
It is not allowed to set both pts_increment and pts_percentage at the same time.
Due to the MPI message size limit of \(2^{31}-1\) elements the number of time-series must not exceed that value.
radius#
- Fortran Type: R (10)
- Default: 0.0
-
SI-Unit: m
Particle radius.
The particle radius steers the viscous friction of particles and must be assigned if density_ratio /= 0.0.
radius can be assigned a different value for each particle group (see number_of_particle_groups). If the number of values given for radius is less than the number of groups defined by number_of_particle_groups), then the last given value is used for all remaining groups.
radius_classes#
- Fortran Type: I
-
Default: 20
Number of radius classes to be used in the collision efficiency table.
This parameter comes into effect if a fast method is used for the collision_kernel (flagged as '_fast'). It defines the number of radius classes which spawn the collision-efficiency table. The interval [10-6, 0.0002] m is divided into n = radius_classes logarithmic equidistant parts.
radius_merge#
- Fortran Type: R
- Default: 1e-07
-
SI-Unit: m
Radius threshold for merging of superdroplets.
Only superdroplets with a radius smaller than radius_merge are considered for merging.
This parameter is only effective if merging = .T..
radius_split#
- Fortran Type: R
- Default: 4e-05
-
SI-Unit: m
Radius threshold for splitting of superdroplets.
Only superdroplets with a radius larger than radius_split are considered for splitting. The default value of 40 µm is found to give reasonable results of the rain-drop distribution in simulations of shallow cumulus clouds. It may need adjustment for different applications.
This parameter is only effective if splitting = .T..
random_start_position#
- Fortran Type: L
-
Default: .FALSE.
Randomize the initial particle position within certain limits.
By default, the initial positions of particles within a source are exactly defined by pst, psl, psr, pss, psn, psb, pdx, pdy, and pdz. With random_start_position = .T., the initial positions of the particles are randomly offset from these positions within certain limits.
read_particles_from_restartfile#
- Fortran Type: L
-
Default: .TRUE.
Read particle data from the previous run.
By default, restart data for particles are read from file PARTICLE_RESTART_DATA_IN, which is created by the preceding run. If particle restart data should not be read, or if the preceding run had no particle transport activated (see particle_advection_start), then read_particles_from_restartfile = .F. is required.
rm#
- Fortran Type: R (3)
- Default: Depends on aero_type
-
SI-Unit: m
Mode radius of the log-normal distribution steering the initial dry aerosol spectrum.
Up to three values of rm can be prescribed. See aero_type for more details.
seed_follows_topography#
- Fortran Type: L
-
Default: .FALSE.
Heights of initial particles are interpreted relative to the given topography.
If seed_follows_topography = .F., the vertical borders of the particle sources psb and pst are interpreted to be above the domain origin. If seed_follows_topography = .T., psb and pst are interpreted to be relative to the given topography.
splitting#
- Fortran Type: L
-
Default: .FALSE.
Switch on/off the splitting algorithm for cloud droplets.
This parameter is only effective if cloud_droplets = .T..
The splitting algorithm splits particles which fulfill certain criterions into several superdroplets with a reduced number of represented particles of every superdroplet (i.e. a reduced weighting factor). The mechanism allows an improved representation of the right tail of the drop size distribution with a feasible amount of computational costs. The splitting algorithm is executed at every timestep.
The splitting algorithm can be steered by the following parameters: max_number_particles_per_gridbox, radius_split, splitting_factor, splitting_factor_max, splitting_function, splitting_mode, and weight_factor_split.
Note: Splitting of particles results in an increase of computational resources. More particles must be calculated (increase in CPU time) and stored (increase in memory demand). Therefore, the limits of the splitting algorithm should be choosen very carefully.
splitting_factor#
- Fortran Type: I
-
Default: 2
Number of particles created when splitting a single particle.
This parameter is used for splitting_mode = 'const'.
If a superdroplet is splitted,
splitting_factor - 1new particles are created with all having the same properties as the original superdroplet. The weighting factor of each of the superdroplets is divided by the splitting_factor.For splitting_mode = 'cl_av' and 'gb_av', splitting_factor is calculated automatically and given by the ratio of the actual weighting factor of a superdroplet and the approximated number concentration (for cloud droplets with a similar radius in a grid volume).
splitting_factor_max#
- Fortran Type: I
-
Default: 5
Upper bound of splitting_factor if splitting_factor is calculated automatically.
This parameter is used for splitting_mode = 'cl_av' and 'gb_av' where the splitting_factor is calculated automatically and can get very large values which are not feasible.
splitting_function#
- Fortran Type: C*5
-
Default: gamma
Function used to approximate the drop-size distribution.
This parameter is used for splitting_mode = 'cl_av' and 'gb_av'.
Currently 3 choices are available:
-
exp
An exponential distribution.
-
gamma
A gamma size distribution.
-
log
A log-normal distribution.
-
splitting_mode#
- Fortran Type: C*5
-
Default: const
Mode used for the splitting algorithm.
Currently 3 choices are available:
-
const
Use a constant splitting_factor when splitting superdroplets.
Superdroplets having a radius \(\geq\) radius_split and a weighting factor \(\geq\) weight_factor_split are split into splitting_factor superdroplets. The weighting factor of the superdroplet and all created clones is reduced by splitting_factor.
-
cl_av
Use a critical radius radius_split and a splitting function splitting_function to steer the splitting of superdroplets.
The critical weighting factor is calculated while using an approximation of the drop size distribution based on the given splitting_function. The drop-size distribution (discretized in 100 bins in a range from 1.0 µm to 1 mm) is calculated as an average over all cloudy grid boxes. Superdroplets which have a larger radius and larger weighting factor are split into several superdroplets. In this case the splitting factor is calculated from the weighting factor of the superdroplet and the approximated number concentration for droplets of such a size. The weighting factor of the superdroplet and all created clones is updated accordingly.
-
gb_av
Same as for splitting_mode = 'cl_av' but a drop-size distribution is calculated for every grid box.
-
step_dealloc#
- Fortran Type: I
-
Default: 100
Number of timesteps after which particle memory is deallocated.
The parameter is only effective if deallocate_memory = .T..
If the number of particles in a grid box exceeds the allocated memory, new memory is allocated. However, in case the number of particles per grid box is only temporarily high, most of the memory remains unused.
If deallocate_memory = .T., the allocated memory used for particles will be dynamically adjusted with respect to the current number of particles after step_dealloc timesetps.
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Switch off the particle module.
If set to .T., the particle module is not activated even if the namelist is present in the parameter file.
unlimited_dimension#
- Fortran Type: L
-
Default: .TRUE.
Set the time dimension of the netCDF output file to be unlimited.
use_sgs_for_particles#
- Fortran Type: L
-
Default: .FALSE.
Use subgrid-scale (SGS) velocities for particle advection.
These velocities are calculated from the resolved and subgrid-scale TKE using the Monte-Carlo random-walk method described by Weil et al. (2004). When using this method, the time step for the advancement of the particles is limited by the so-called Lagrangian time scale. This may be smaller than the current LES time step so that several particle (sub-)time steps have to be carried out within a single LES time step.
In order to limit the number of sub-time steps (and to limit the CPU-time), the minimum value for the particle time step is defined by dt_min_part.
Please see also the discussion in ticket 1489 concerning the impact of SGS speeds on particle spreading.
vertical_particle_advection#
- Fortran Type: L (10)
-
Default: .TRUE.
Switch on/off vertical particle transport.
By default, particles are transported along all three directions in space. With vertical_particle_advection = .F., the particles will only be transported horizontally.
vertical_particle_advection can be set different for each particle group (see number_of_particle_groups).
weight_factor_merge#
- Fortran Type: R
-
Default: Depends on initial_weighting_factor
Threshold for merging of superdroplets.
By default, weight_factor_merge = 0.5 initial_weighting_factor.
Only superdroplets with a weighting factor \(\leq\) weight_factor_merge will be merged.
weight_factor_split#
- Fortran Type: R
-
Default: Depends on initial_weighting_factor
Threshold for splitting of superdroplets.
The parameter is only effective if splitting_mode = 'const'.
By default, weight_factor_split = 0.1 initial_weighting_factor.
Superdroplets with a weighting factor \(\geq\) weight_factor_split are considered for cloning.
write_particle_statistics#
- Fortran Type: L
-
Default: .FALSE.
Switch on/off output of particle statistics.
If write_particle_statistics = .T., statistical information are output to file PARTICLE_INFOS by each PE. These statistics may be used for debugging. The statistics contain
- time step count,
- simulated time,
- total number of particles used,
- number of particles exchanged between neighboring PEs.
Note: For parallel runs files may become very large and performance of PALM may decrease.
Plant Canopy Parameters#
alpha_lad#
- Fortran Type: R
-
Default: undefined
Alpha coefficient for leaf area density calculation.
Dimensionless coefficient required for constructing the leaf-area density (LAD) profile using the beta probability density function (following Markkanen et al., 2003):
\[ f_\text{PDF}(\frac{z}{H},\alpha,\beta) = \frac{(\frac{z}{H})^{\alpha-1}\;(1-\frac{z}{H})^{\beta-1}}{\int_{0}^{1}\;(\frac{z}{H})^{\alpha-1}\;(1-\frac{z}{H})^{\beta-1}\;d(\frac{z}{H})} \,\, , \]where \(z\) is the height above ground, \(H\) is the canopy height, and \(\alpha\) (alpha_lad) and \(\beta\) (beta_lad) are the coefficients to be prescribed. The actual LAD are calculated as
\[ \text{LAD}(z,\alpha,\beta) = \text{LAI} \cdot f_\text{PDF}(\frac{z}{H},\alpha,\beta) \cdot H \,\, , \]with the leaf-area index (LAI, defined as the vertical integral of LAD) prescribed by lai_beta.
lai_beta must be set to a non-zero value in order to use the beta probability density function for the LAD-profile construction. alpha_lad and beta_lad steer the vertical distribution of leaf area within the canopy volume. Values for alpha_lad must be greater than zero. Increasing alpha_lad moves the leaf area toward the top of the canopy. If alpha_lad is given, beta_lad must be given, too.
The LAD profile can also be constructed by prescribing vertical gradients of LAD (lad_vertical_gradient, lad_vertical_gradient_level), starting from the prescribed surface value lad_surface.
beta_lad#
- Fortran Type: R
-
Default: undefined
Beta coefficient for leaf area density calculation.
Dimensionless coefficient required for constructing the leaf-area density (LAD) profile, using a beta probability density function (see alpha_lad for details).
beta_lad and alpha_lad steer the vertical distribution of leaf area within the canopy volume. Values for beta_lad must be greater than zero. Increasing beta_lad moves the leaf area toward the floor of the canopy. If beta_lad is given, alpha_lad must be given, too.
The LAD profile can also be constructed by prescribing vertical gradients of LAD (lad_vertical_gradient, lad_vertical_gradient_level), starting from the prescribed surface value lad_surface.
canopy_drag_coeff#
- Fortran Type: R
-
Default: 0.0
Drag coefficient used in the plant-canopy model.
The drag coefficient is used to calculate the drag force due to the plant canopy. Please refer to the documentation of the plant-canopy model for further details.
canopy_drag_coeff must be greater than zero. Typical values are 0.15 (Shaw and Schumann, 1992) or 0.20 (Cescatti and Marcolla, 2004).
canopy_mode#
- Fortran Type: C*30
-
Default: homogeneous
Set up mode for the canopy layer.
Currently 3 choices are available:
-
homogeneous
Horizontally homogeneous plant canopy extending over the entire model domain.
-
read_from_file
Requires a static driver (file suffix
_static) with 3D information of leaf area density (see LAD definition in the static driver). -
user_defined
According to user settings in subroutine user_init_plant_canopy.f90.
-
cthf#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: W m-2
Average heat flux at the top of the plant canopy.
This parameter prescribes a heat flux at the top of the plant canopy (canopy top heat flux). It is assumed that solar radiation penetrates the canopy and warms the foliage which, in turn, warms the air in contact with it. Based on cthf, the heat fluxes inside the canopy down to the canopy floor are determined by a decaying exponential function that is dependent on the cumulative leaf area index (Shaw and Schumann, 1992).
In areas with canopy, cthf overrides surface_heatflux.
lad_surface#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: m2 m-3
Surface value of the leaf-area density (LAD).
This parameter assigns the value of LAD at the surface (
k = 0). Starting from this value, the LAD profile is constructed via lad_vertical_gradient and lad_vertical_gradient_level.The LAD profile can also be constructed using a beta probability density function by prescribing values for parameters alpha_lad, beta_lad, and lai_beta. In this case, lad_surface must not be set.
lad_type_coef#
- Fortran Type: R (10)
-
Default: 1.0
Multiplicative coefficient for different plant-canopy types.
This parameter is only active when data is read from ASCII file PLANT_CANOPY_DATA_3D.
lad_vertical_gradient#
- Fortran Type: R (10)
- Default: 0.0
-
SI-Unit: m2 m-4
Gradients of the leaf area density.
The leaf-area density (LAD) gradient holds starting from the height level defined by lad_vertical_gradient_level up to the level defined by pch_index; precisely: for all uv levels
kwherezu(k) > lad_vertical_gradient_levelandk <= pch_index, LAD is set aslad(k) = lad(k-1) + dzu(k) * lad_vertical_gradientAbove pch_index, LAD will be 0.0.
A total of 10 different gradients for 11 height intervals (10 intervals if lad_vertical_gradient_level(1) = 0.0) can be assigned. The LAD at the surface is assigned via lad_surface.
lad_vertical_gradient_level#
- Fortran Type: R (10)
- Default: 0.0
-
SI-Unit: m
Height levels for constructing the vertical LAD profile.
Height level from which the gradient of the leaf-area density (LAD), defined by lad_vertical_gradient, is effective.
The height levels must be assigned in ascending order. The default values result in an LAD that is constant with height up to the top of the plant canopy layer defined by pch_index. For the piecewise linear construction of an LAD profile see lad_vertical_gradient.
lai_beta#
- Fortran Type: R
-
Default: 0.0
Leaf-area index (LAI) used to construct a vertical leaf-area density (LAD) profile.
lai_beta is used as a parameter of the beta probability density function to construct the LAD profile. See alpha_lad for details.
lai_beta must be set to a non-zero value, and parameters alpha_lad and beta_lad must be given, too.
leaf_scalar_exch_coeff#
- Fortran Type: R
-
Default: 0.0
Scalar exchange coefficient for a "bulk" leaf.
The parameter is only relevant if passive_scalar = .T..
The value of the scalar exchange coefficient is required for the parametrization of the sources and sinks of scalar concentration due to the canopy.
leaf_surface_conc#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: kg m-3 or ppm
Scalar concentration at the leaf surface.
Concentration of a passive scalar at the surface of a "bulk" leaf. Its unit depends if the scalar is treated as particles (kg m-3) or as a gas (ppm).
The parameter is only relevant if passive_scalar = .T..
leaf_surface_conc is required for the parametrization of the sources and sinks of scalar concentration due to the canopy.
pch_index#
- Fortran Type: I
-
Default: 0
Grid-point index of the upper boundary of the plant-canopy layer.
Above pch_index the leaf-area density (LAD) is automatically set to zero. Up to pch_index an LAD profile can be prescribed in two possible ways:
-
Creating a piecewise linear LAD profile by prescribing the parameters lad_surface, lad_vertical_gradient and lad_vertical_gradient_level.
-
Employing a beta probability density function for the vertical leaf-area distribution by prescribing coefficients alpha_lad, beta_lad and lai_beta (see, e.g., Markkanen et al. (2003)).
pch_index is always relative to the topography, e.g. if the topography surface at a specific point
(i,j)is atk= 10, and pch_index = 5, the canopy at this point extends fromk= 10 tok= 15.If the LAD profile is given via an input file, parameter pch_index is automatically set according to the information given in the input file (number of vegetation layers - 1).
-
plant_canopy_transpiration#
- Fortran Type: L
-
Default: .FALSE.
Turn on evapotranspiration and corresponding latent heat flux from resolved plant canopy.
The calculation utilizes short-wave and long-wave radiation fluxes calculated in the radiation transfer model.
The calculation of transpiration rate is based on the Jarvis-Stewart model with parametrizations described in Daudet et al. (1999) and Ngao, Adam and Saudreau (2017) with some modifications according to Stewart (1988).
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Switch to turn on/off the module.
If switch_off_module = .T., the plant-canopy module is not activated even if the namelist is present in the parameter file.
Radiation Parameters#
albedo#
- Fortran Type: R
-
Default: undefined
Surface albedo.
A value for this parameter should be assigned when a broadband albedo value is required for the whole domain and no describtion of the albedo_type is given via static driver, i.e. albedo_type = 0
This parameter will be overwritten when albedo data is given via albedo_type.
albedo_lw_dif#
- Fortran Type: R
-
Default: undefined
Surface albedo for longwave diffuse radiation for a solar angle of 60°.
This parameter is used when radiation_scheme is set to 'rrtmg' or 'tenstream'. A value for this parameter must be assigned if albedo_type = 0.
albedo_lw_dir#
- Fortran Type: R
-
Default: undefined
Surface albedo for longwave direct radiation for a solar angle of 60°.
This parameter is used when radiation_scheme is set to 'rrtmg' or 'tenstream'. A value for this parameter must be assigned if albedo_type = 0.
albedo_sw_dif#
- Fortran Type: R
-
Default: undefined
Surface albedo for shortwave direct radiation for a solar angle of 60°.
This parameter is used when radiation_scheme is set to 'rrtmg' or 'tenstream'. A value for this parameter must be assigned if albedo_type = 0.
albedo_sw_dir#
- Fortran Type: R
-
Default: undefined
Surface albedo for shortwave direct radiation for a solar angle of 60°.
This parameter is used when radiation_scheme is set to 'rrtmg' or 'tenstream'. A value for this parameter must be assigned if albedo_type = 0.
albedo_type#
- Fortran Type: I
-
Default: 5
Albedo type of the surface.
This parameter controls the choice of the surface albedos for direct/diffuse/broadband radiation according to the following list of predefined land surfaces.
For radiation_scheme = 'rrtmg' the shortwave and longwave albedo values are used and the actual albedos are then calculated based on the current solar angle after Briegleb et al. (1986) and Briegleb (1992). The displayed values are given for a solar zenith angle of 60° and will vary in the course of the day according to Briegleb et al. (1986) and Briegleb (1992). Setting one or more of the parameters albedo_lw_dif, albedo_lw_dir, albedo_sw_dif, and/or albedo_sw_dir will overwrite the respective values set by albedo_type. By default, albedo_lw_dif = albedo_lw_dir and albedo_sw_dif = albedo_sw_dir. Moreover, constant_albedo = .T. will keep the albedos constant during the simulation.
For radiation_scheme = 'clear-sky' the broadband albedo is used and does not vary in the course of the simulation.
bufsize_alltoall#
- Fortran Type: I
-
Default: 0
Maximum number of items sent in
MPI_AlltoAllcalls at once (0 = infinite).Some implementations of the MPI protocol or their underlying hardware have limits on the size of sending/receiving buffers. If the user experiences problems in
MPI_AlltoAlltransfer, e.g. segmentation faults in MPI, setting this to a value different than zero replaces the large transfers by sequences of limited-size exchanges. The default setting 0 means no limit, which is suitable for most situations and it should not be changed unless there are problems with MPI. The optimal value for best performance highly depends on the parallel hardware and its configuration and needs to be determined by testing; a good starting point is 1.000.000. The value should not be set lower than approx. 1000 as too many small exchanges could lead to severely degraded performance.
constant_albedo#
- Fortran Type: L
-
Default: .FALSE.
Flag to keep the surface albedo constant during the simulation.
If set to .T., the surface albedos are kept constant during the entire simulation, while if set to .F., the actual albedos are calculated after Briegleb et al. (1986) and Briegleb (1992).
dt_radiation#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: s
Time step of the radiation model.
This parameter describes how frequently the radiation fluxes are updated. This parameter must be set in the namelist, because the default value will issue an error message and abort the simulation, otherwise.
emissivity#
- Fortran Type: R
-
Default: 0.95
Surface emissivity (0.0 - 1.0) for user-defined grid points.
Please note, by default emissivity is determined by the prescribed vegetation-, pavement-, water-, or building type at the given surface element. Setting this parameter has only an effect if vegetation-, pavement-, water-, or building type is set to zero, i.e. a user-defined value.
At the moment, only a single emissivity value can be used for each band in the atmospheric window.
lw_radiation#
- Fortran Type: L
-
Default: .TRUE.
Flag to switch the calculation of longwave radiation on.
This flag is effective only when radiation_scheme is set to 'rrtmg' or 'tenstream'.
mrt_geom#
- Fortran Type: I
-
Default: 1
Method for MRT direction weights simulating a sphere or a human body.
Currently 3 choices are available:
-
0
Sphere (no weighting).
-
1
Original weighting function simulating human body.
-
2
Human body shape simulated as ellipsoid.
-
mrt_geom_params#
- Fortran Type: R (2)
-
Default: [0.12, 0.88]
Parameters of the selected weighting method (mrt_geom).
For mrt_geom = 1 and mrt_geom = 2, mrt_geom_params represents width and height of the human body.
mrt_include_sw#
- Fortran Type: L
-
Default: .TRUE.
Flag to include shortwave radiation into the mean radiant temperature calculation.
This flag should be set to .T. when shortwave radiant flux for MRT boxes (rtm_mrt_sw) is required in the output.
mrt_minlevel#
- Fortran Type: I
-
Default: 0
Lowest vertical box (grid index) above surface for which to calculate MRT.
mrt_nlevels#
- Fortran Type: I
-
Default: 0
Number of vertical boxes above surface for which to calculate mean radiant temperature.
This value is used also for calculation of MRT and PET values in the biometeorology module (bio_mrt and bio_pet output variables).
mrt_skip_roof#
- Fortran Type: L
-
Default: .TRUE.
Flag to skip calculating mean radiant temperature above the roof surfaces.
This flag limits the mean radiant temperature calculations to the levels above the ground.
net_radiation#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: Wm-2
Net radiation flux at the surface.
This parameter is only used in case of radiation_scheme = 'constant'.
nrefsteps#
- Fortran Type: I
-
Default: 3
Number of reflection steps to be performed inside the RTM.
The number of reflection steps for the reflected short- and long-wave radiation between mutually visible surfaces.
plant_lw_interact#
- Fortran Type: L
-
Default: .TRUE.
Flag to allow plant canopy to interact with longwave radiation.
When the flag is set to .T., it enables absorption and emission of longwave radiation by the resolved plant canopy. If value is set to .F., the plant canopy is transparent to longwave radiation.
localized_raytracing#
- Fortran Type: L
-
Default: .FALSE.
Flag to enable the localized parallelization scheme for raytracing in RTM.
The new localized parallelization scheme uses different distribution of workload and data transfer while raytracing. This scheme does not need one-sided MPI operations (see raytrace_mpi_rma) and it does not use several global arrays, thus improving scalability. It is also significantly faster. Currently it has to be enabled manually, but it will later become the default scheme.
radiation_interactions_on#
- Fortran Type: L
-
Default: .TRUE.
Flag to deactivate RTM even if vertical urban/land surfaces or trees exist in the domain.
Setting this parameter to .F. deactivates the RTM even if vertical urban/land surfaces or trees exist in the domain. In this case, all surfaces will receive radiation fluxes directly from the choosen radiation model (e.g. 'rrtmg' or 'clear-sky').
Warning: radiation_interactions_on = .T. is not allowed, if the bulk cloud model (BCM) is used together with the urban- and/or land-surface model (USM and/or LSM) and the radiation model.
radiation_only#
- Fortran Type: L
-
Default: .FALSE.
Flag to activate the radiation model without urban/land surfaces being defined.
Setting this parameter to .T. activates the radiation model without urban/land surfaces being defined. In such radiation-only simulations, the radiative transfer equation is only solved for the atmosphere and no energy balance is solved at the surface. The surface parameter albedo and emissivity are either loaded by the namelist (albedo_type, albedo, emissivity) or read by a static driver file.
Warning: Setting this flag to .T. is not allowed in combination with the urban- and/or land-surface model (USM and/or LSM) or radiation_interactions_on = .T.. Furthermore, it is only implemented for radiation_scheme = 'rrtmg'.
radiation_scheme#
- Fortran Type: C*10
-
Default: clear-sky
Radiation scheme to be used.
Currently 5 choices are available:
-
constant
A constant net radiation at the surface is prescribed (see net_radiation).
-
clear-sky
A simple clear sky model is used to calculate the radiation fluxes at the surface (shortwave incoming, shortwave outgoing, longwave incoming, longwave outgoing) and, consequently, the net radiation at the surface. This scheme requires setting albedo, origin_date_time, longitude and latitude.
-
external
External radiative input of short- and long-wave downwelling radiation is used, which can be taken from observations or mesoscale models. Note, in the case of a nested run, each child model will read the radiation input either from its own dynamic input file, or if a dynamic input file is not available for each of the child models, it will be read from the dynamic input file for the root model.
External radiation input can be provided as
level-of-detail = 1(radiation depends only on time and is horizontally homogeneous) or aslevel-of-detail = 2(radiation depends on time, y, x and can be horizontally heterogeneous). Please note, at the momentlevel-of-detail = 2is not possible in combination with buildings or complex orography where surface reflections occur. -
rrtmg
The RRTMG model is used to calculate the radiative heating rates for each model column. This scheme requires setting origin_date_time, longitude and latitude. The following parameters can be set optionally: albedo_type (and/or albedo_lw_dif, albedo_lw_dir, albedo_sw_dif, albedo_sw_dir). However, constant_albedo can be used to fix the albedo during the simulation. It is also possible to use the switches lw_radiation and sw_radiation.
Please note that the use of RRTMG requires the user to compile PALM with
-D__rrtmgpreprocessor directive (in the.palm.config.<configuration_identifier>file). But before this, the RRTMG libraries must be installed, which is usually done automatically by the PALMinstaller. -
tenstream
The TenStream model is used to calculate the full 3-D radiative transfer equation. Unlike RRTMG, it approximately solves the radiative transfer equation in 3-D and computes irradiance and heating rates from optical properties. In addition to the up- and down-ward fluxes, the TenStream solver computes the radiative transfer coefficients for sideward streams.
-
radiation_start_time#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: s
Time after which the radiation model is switched on.
This parameter can be used to allow the LES model to develop turbulence before the flow is affected by radiation.
radiation_volumetric_flux#
- Fortran Type: L
-
Default: .FALSE.
Flag indicating whether volumetric radiative fluxes will be calculated.
raytrace_discrete_azims#
- Fortran Type: I
-
Default: 80
Number of horizontal discrete directions (azimuths) for angular discretization.
This parameter is used in the RTM model to calculate the sky view factor, surface-surface view factor and direct solar visibility for all surfaces.
raytrace_discrete_elevs#
- Fortran Type: I
-
Default: 40
Number of vertical discrete elevations for angular discretization.
This parameter is used to calculate the sky view factor, surface-surface view factor and direct solar visibility for all surfaces.
raytrace_mpi_rma#
- Fortran Type: L
-
Default: .TRUE.
Flag to enable one-sided MPI communication to access LAD array and grid surfaces for raytracing.
This flag is only for debugging purposes and should not be switched off in real simulations.
Attention: Some machine related settings may be necessary to avoid system degradation when using MPI-RMA communication. See for example http://palm.muk.uni-hannover.de/trac/wiki/doc/app/machine/hlrnIII.
surface_reflections#
- Fortran Type: L
-
Default: .TRUE.
Flag to switch off the surface-surface reflections in RTM.
This flag is intended only for special purposes (e.g. debugging, testing of sensitivities) and should not be switched off for real simulations.
sw_radiation#
- Fortran Type: L
-
Default: .TRUE.
Parameter to switch off the calculation of shortwave radiation.
When using RRTMG, shortwave radiation calls can be switched off by setting sw_radiation = .F.. Note that shortwave radiation is automatically switched off during nighttime.
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Flag to switch off the radiation module.
Setting this flag to .T. will switch off the radiation module although the respective module namelist appears in the namelist file.
trace_fluxes_above#
- Fortran Type: R
-
Default: -1.0
This parameter enables tracing of values of radiative fluxes in RTM.
If this threshold is exceeded, RTM prints a debug message with the maximal value of the corresponding flux with additional useful information (location of maximum, corresponding albedo and emissivity,...). The value 0.0 means to print maxima at all timesteps for all particular radiative fluxes while the value -1.0 (default) disables this check completely.
ts_icollapse#
- Fortran Type: I
-
Default: -1
Flag to return flux results from the background atmosphere.
Currently 2 choices are available:
-
1
Receive the TenStream flux results from the background atmosphere above the dynamical grid.
-
-1
Collapse the TenStream flux results from the background atmosphere above the dynamical grid.
-
unscheduled_radiation_calls#
- Fortran Type: L
-
Default: .FALSE.
Parameter to allow additional calls of the radiation code.
Usually the radiation is called in time intervals as given by dt_radiation, however in the case of fast changes in the skin temperature, it is recommended to update the radiative fluxes independently from the prescribed radiation call frequency to prevent numerical oscillations. The value of the temperature threshold used is 0.2 K.
use_broadband_albedo#
- Fortran Type: L
-
Default: .FALSE.
Flag to use broadband albedo instead of diffuse/direct albedo.
This flag is used only when the radiation scheme RRTMG or TenStream is used.
Salsa Parameters#
advect_particle_water#
- Fortran Type: L
-
Default: .TRUE.
Parameter to switch on the advection of condensed water in aerosol particles.
If advect_particle_water = .F., the aerosol particle water content is calculated at each dt_salsa based on the equilibrium solution using the ZSR method (Stokes and Robinson, 1966).
aerosol_flux_dpg#
- Fortran Type: R (7)
- Default: [1.3e-08, 5.4e-08, 8.6e-07, 2e-07, 2e-07, 2e-07, 2e-07]
-
SI-Unit: m
The number geometric mean diameter per aerosol mode for the aerosol emission.
A total of 7 different aerosol modes can be applied. Example aerosol modes are 'nucleation', 'Aitken', 'accumulation' and 'coarse'.
Used to construct the aerosol emissions if horizontally uniform (salsa_emission_mode = 'uniform' ) or parameterized (salsa_emission_mode = 'parameterized' ) aerosol emissions are applied. Then the aerosol number emission is described by parameters aerosol_flux_dpg, aerosol_flux_sigmag, aerosol_flux_mass_fracs_a, and surface_aerosol_flux.
aerosol_flux_mass_fracs_a#
- Fortran Type: R (7)
-
Default: [1.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0]
Mass fractions of soluble chemical components (subrange 2a) for the horizontally homogeneous aerosol emission (salsa_emission_mode = 'uniform' ).
Given in the same order as the list of activated aerosol chemical components listspec. E.g.
listspec = 'OC', 'NH', 'BC', ' ', ' ', ' ', ' ', aerosol_flux_mass_fracs_a = 0.3, 0.1, 0.6, 0.0, 0.0, 0.0, 0.0,
implies that the mass composition of the aerosol emission is 30% organic carbon, 10% ammonia and 60% black carbon.
Used to construct the aerosol emissions if horizontally uniform (salsa_emission_mode = 'uniform' ) or parameterized (salsa_emission_mode = 'parameterized' ) aerosol emissions are applied. Then the aerosol number emission is described by parameters aerosol_flux_dpg, aerosol_flux_sigmag, aerosol_flux_mass_fracs_a, and surface_aerosol_flux.
Note that
aerosol_flux_mass_fracs_bis not currently implemented. Hence, aerosol emissions are always soluble when using salsa_emission_mode = 'uniform'.
aerosol_flux_sigmag#
- Fortran Type: R (7)
-
Default: [1.8, 2.16, 2.21, 2.0, 2.0, 2.0, 2.0]
The standard deviation of the log-normal aerosol number size distribution per aerosol mode for the aerosol emission.
A total of 7 different aerosol modes can be applied. Example aerosol modes are 'nucleation', 'Aitken', 'accumulation' and 'coarse'.
Used to construct the aerosol emissions if horizontally uniform (salsa_emission_mode = 'uniform' ) or parameterized (salsa_emission_mode = 'parameterized' ) aerosol emissions are applied. Then the aerosol number emission is described by parameters aerosol_flux_dpg, aerosol_flux_sigmag, aerosol_flux_mass_fracs_a, and surface_aerosol_flux.
bc_aer_b#
- Fortran Type: C*20
-
Default: neumann
The bottom boundary condition of the aerosol concentrations.
The same condition applies also for gases if the chemistry module is not applied. If any surface emissions of aerosols and gases are applied (see salsa_emission_mode), bc_aer_b = 'neumann' is required.
Currently 2 choices are available:
-
dirichlet
Constant surface concentration over the entire simulation.
-
neumann
Zero concentration gradient.
-
bc_aer_l#
- Fortran Type: C*20
-
Default: Value of (bc_lr)
The left boundary condition of the aerosol concentrations.
The same condition applies also for gases if the chemistry module is not applied. Horizontal boundary conditions for aerosols can be set independently from horizontal boundary conditions that have been chosen for the remaining model quantities via parameters bc_lr and bc_ns, e.g. you can choose cyclic boundary conditions for aerosols, while the flow field uses non-cyclic conditions, or vice versa.
Currently 3 choices are available:
-
dirichlet
Constant concentration over the entire simulation as given by the initial profiles.
-
neumann
Zero concentration gradient.
-
cyclic
Cyclic along
x. For this value, bc_aer_r = 'cyclic' is required, too.
-
bc_aer_n#
- Fortran Type: C*20
-
Default: Value of (bc_ns)
The north boundary condition of the aerosol concentrations.
The same condition applies also for gases if the chemistry module is not applied. Horizontal boundary conditions for aerosols can be set independently from horizontal boundary conditions that have been chosen for the remaining model quantities via parameters bc_lr and bc_ns, e.g. you can choose cyclic boundary conditions for aerosols, while the flow field uses non-cyclic conditions, or vice versa.
Currently 3 choices are available:
-
dirichlet
Constant concentration over the entire simulation as given by the initial profiles.
-
neumann
Zero concentration gradient.
-
cyclic
Cyclic along
y. For this value, bc_aer_s = 'cyclic' is required, too.
-
bc_aer_r#
- Fortran Type: C*20
-
Default: Value of (bc_lr)
The right boundary condition of the aerosol concentrations.
The same condition applies also for gases if the chemistry module is not applied. Horizontal boundary conditions for aerosols can be set independently from horizontal boundary conditions that have been chosen for the remaining model quantities via parameters bc_lr and bc_ns, e.g. you can choose cyclic boundary conditions for aerosols, while the flow field uses non-cyclic conditions, or vice versa.
Currently 3 choices are available:
-
dirichlet
Constant concentration over the entire simulation as given by the initial profiles.
-
neumann
Zero concentration gradient.
-
cyclic
Cyclic along
x. For this value, bc_aer_l = 'cyclic' is required, too.
-
bc_aer_s#
- Fortran Type: C*20
-
Default: Value of (bc_ns)
The south boundary condition of the aerosol concentrations.
The same condition applies also for gases if the chemistry module is not applied. Horizontal boundary conditions for aerosols can be set independently from horizontal boundary conditions that have been chosen for the remaining model quantities via parameters bc_lr and bc_ns, e.g. you can choose cyclic boundary conditions for aerosols, while the flow field uses non-cyclic conditions, or vice versa.
Currently 3 choices are available:
-
dirichlet
Constant concentration over the entire simulation as given by the initial profiles.
-
neumann
Zero concentration gradient.
-
cyclic
Cyclic along
y. For this value, bc_aer_n = 'cyclic' is required, too.
-
bc_aer_t#
- Fortran Type: C*20
-
Default: neumann
The top boundary condition of the aerosol concentrations.
The same condition applies also for gases if the chemistry module is not applied.
Currently 3 choices are available:
-
dirichlet
Constant concentration over the entire simulation.
-
neumann
Zero concentration gradient.
-
initial_gradient
Gradient is calculated from the initial concentration profile.
-
depo_pcm_par#
- Fortran Type: C*20
-
Default: zhang2001
Method to solve the aerosol size specific dry deposition velocity.
The surface material is specified via depo_pcm_type.
Currently 2 choices are available:
-
zhang2001
See Zhang et al. (2001).
-
petroff2010
-
depo_pcm_type#
- Fortran Type: C*20
-
Default: deciduous_broadleaf
Leaf type applied in the dry deposition model.
Currently 4 choices are available:
-
deciduous_broadleaf
-
deciduous_needleleaf
-
evergreen_broadleaf
-
evergreen_needleleaf
-
depo_surf_par#
- Fortran Type: C*20
-
Default: zhang2001
Method to solve the dry deposition velocity for aerosols over both horizontal and vertical surfaces.
The land use type applied in the parametrisations are imported from the land and urban surface modules. If the surfaces are not specified using the urban surface or land surface module, the land use type urban is applied for all surfaces.
Currently 2 choices are available:
-
zhang2001
See Zhang et al. (2001).
-
petroff2010
-
dpg#
- Fortran Type: R (7)
- Default: [1.3e-08, 5.4e-08, 8.6e-07, 2e-07, 2e-07, 2e-07, 2e-07]
-
SI-Unit: m
The number geometric mean diameter per aerosol mode.
A total of 7 different aerosol modes can be applied. Example aerosol modes are 'nucleation', 'Aitken', 'accumulation' and 'coarse'. In case of initializing_actions = 'set_constant_profiles', the initial aerosol size distribution is described by dpg, sigmag, and n_lognorm.
dt_salsa#
- Fortran Type: R
- Default: 0.1
-
SI-Unit: s
The time step for calling aerosol dynamic processes of SALSA.
For switching on individual processes, see nlcnd, nlcndgas, nlcndh2oae, nlcoag, nldepo, nldepo_pcm, nldepo_surf, nldistupdate, and nsnucl.
emiss_factor_main#
- Fortran Type: R
-
Default: 0.0
Constant emission scaling factor for main street types.
Used if salsa_emission_mode = 'parameterized'.
emiss_factor_side#
- Fortran Type: R
-
Default: 0.0
Constant emission scaling factor for side street types.
Used if salsa_emission_mode = 'parameterized'.
feedback_to_palm#
- Fortran Type: L
-
Default: .FALSE.
Parameter to switch on the dynamic feedback to the flow due to condensation of water vapor on aerosol particles.
In case of feedback_to_palm = .F., the salsa module does not interact with the flow.
h2so4_init#
- Fortran Type: R
- Default: 1.0
-
SI-Unit: m-3
Initial number concentration of gaseous sulphuric acid H2SO4.
hno3_init#
- Fortran Type: R
- Default: 1.0
-
SI-Unit: m-3
Initial number concentration of gaseous nitric acid HNO3.
listspec#
- Fortran Type: C*3 (7)
-
Default: ['SO4', ' ', ' ', ' ', ' ', ' ', ' ']
List of activated aerosol chemical components.
All chemical components included in the simulation must be activated here, e.g.
listspec = 'OC', 'NH', 'BC', ' ', ' ', ' ', ' ',
implies that aerosol particles can contain organic carbon, ammonia and black carbon.
Currently 7 choices are available:
-
SO4
sulphates
-
OC
organic carbon
-
BC
black carbon
-
DU
dust
-
SS
sea salt
-
NH
ammonia
-
NO
nitrates
-
main_street_id#
- Fortran Type: I (99)
-
Default: 0
Index for identifying main streets following street type classes from
OpenStreetMap.Used if salsa_emission_mode = 'parameterized'.
mass_fracs_a#
- Fortran Type: R (7)
-
Default: [1.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0]
Mass fractions of soluble chemical components (subrange 2a).
Given in the same order as the list of activated aerosol chemical components listspec, e.g.
listspec = 'OC', 'NH', 'BC', ' ', ' ', ' ', ' ', mass_fracs_a = 0.3, 0.1, 0.6, 0.0, 0.0, 0.0, 0.0,
implies that the initial mass composition of the aerosol particles is 30% organic carbon, 10% ammonia and 60% black carbon.
mass_fracs_b#
- Fortran Type: R (7)
-
Default: [0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0]
Mass fractions of insoluble chemical components (subrange 2b).
Given in the same order as the list of activated aerosol chemical components listspec.
Setting mass_fracs_b > 0.0 and nf2a < 1.0 allows for the description of externally mixed aerosol particle populations in the subrange 2. However, this notably increases the computational demand.
If SUM(mass_fracs_b) = 0.0, all aerosol particles are assumed to be soluble and the subrange 2b is not initialised.
max_street_id#
- Fortran Type: I (99)
-
Default: 0
Maximum index value for identifying all (main and side) streets following street type classes from
OpenStreetMap.Used if salsa_emission_mode = 'parameterized'.
n_lognorm#
- Fortran Type: R (7)
- Default: [104000000000.0, 32300000000.0, 5400000.0, 0.0, 0.0, 0.0, 0.0]
-
SI-Unit: m-3
The total aerosol number concentration per aerosol mode.
A total of 7 different aerosol modes can be applied. Example aerosol modes are 'nucleation', 'Aitken', 'accumulation' and 'coarse'. In case of initializing_actions = 'set_constant_profiles', the initial aerosol size distribution is described by dpg, sigmag, and n_lognorm.
nbin#
- Fortran Type: I (2)
-
Default: [3, 7]
Number of aerosol size bins per subrange.
nesting_offline_salsa#
- Fortran Type: L
-
Default: .TRUE.
Parameter to switch on/off offline nesting for salsa variables.
If nesting_offline_salsa = .F. , the boundary conditions for salsa variables are defined via parameters bc_aer_b, bc_aer_t, bc_aer_l, bc_aer_r, bc_aer_n, and bc_aer_s.
nf2a#
- Fortran Type: R
-
Default: 1.0
The number fraction allocated to subrange 2a.
The number fraction allocated to the subrange 2b will be then 1.0 - nf2a.
nh3_init#
- Fortran Type: R
- Default: 1.0
-
SI-Unit: m-3
Initial number concentration of gaseous ammonia NH3.
nj3#
- Fortran Type: I
-
Default: 1
Parametrisation for calculating the apparent formation rate of 3 nm sized aerosol particles \(J_3\).
Currently 3 choices are available:
-
1
condensational sink (Kerminen and Kulmala, 2002)
-
2
coagulational sink (Lehtinen et al. 2007)
-
3
coagS+self-coagulation (Anttila et al. 2010)
-
nlcnd#
- Fortran Type: L
-
Default: .FALSE.
Parameter to switch on the condensation of gaseous compounds on aerosol particles.
nlcndgas#
- Fortran Type: L
-
Default: .FALSE.
Parameter to switch on the condensation of gaseous compounds, excluding water vapour, on aerosol particles.
Requires nlcnd = .T..
nlcndgash2oae#
- Fortran Type: L
-
Default: .FALSE.
Parameter to switch on the condensation of water vapour on aerosol particles.
Requires nlcnd = .T..
nlcoag#
- Fortran Type: L
-
Default: .FALSE.
Parameter to switch on the coagulation of aerosol particles.
nldepo#
- Fortran Type: L
-
Default: .FALSE.
Parameter to switch of the dry deposition and sedimentation of aerosol particles.
nldepo_pcm#
- Fortran Type: L
-
Default: .FALSE.
Parameter to switch on aerosol dry deposition on resolved scale vegetation.
The parametrisation to calculate the size-dependent deposition velocity is set by depo_pcm_par. Requires nldepo = .T..
Note that currently the deposition velocity is calculated by default for deciduous broadleaf trees.
nldepo_surf#
- Fortran Type: L
-
Default: .FALSE.
Parameter to switch aerosol dry deposition on topography elements (ground, wall, roofs).
The parametrisation to calculate the size-dependent deposition velocity is set by depo_surf_par. Requires nldepo = .T..
Note that currently the deposition velocity is calculated by default for deciduous broadleaf trees.
nldistupdate#
- Fortran Type: L
-
Default: .TRUE.
Parameter to switch on the aerosol number size distribution update switch.
If nldistupdate = .F., aerosol particles that become too small or large in their size bin are not allowed to move to another size bin.
nsnucl#
- Fortran Type: I
-
Default: 0
The nucleation scheme applied.
Requires nlcnd = .T.. Note that the nucleation schemes were not evaluated in Kurppa et al. (2019).
Currently 10 choices are available:
-
0
nucleation is switched off
-
1
binary nucleation (Vehkamäki et al., 2002)
-
2
activation type nucleation (Riipinen et al., 2007)
-
3
kinetic nucleation (Sihto et al., 2006)
-
4
ternary nucleation (Napari et al., 2002a,b)
-
5
organic nucleation (Paasonen et al., 2010)
-
6
sum of binary and organic nucleation (Paasonen et al., 2010)
-
7
heteromolecular nucleation (Paasonen et al., 2010)
-
8
homomolecular nucleation of H2SO4 and heteromolecular nucleation of H2SO4 and organics (Paasonen et al., 2010)
-
9
homomolecular nucleation of H2SO4 and organics, and heteromolecular nucleation of H2SO4 and organics (Paasonen et al., 2010)
-
ocnv_init#
- Fortran Type: R
- Default: 1.0
-
SI-Unit: m-3
Initial number concentration of gaseous non-volatile organic compounds.
ocsv_init#
- Fortran Type: R
- Default: 1.0
-
SI-Unit: m-3
Initial number concentration of gaseous semi-volatile organic compounds.
reglim#
- Fortran Type: R (3)
- Default: [3e-09, 5e-08, 1e-05]
-
SI-Unit: m
Aerosol diameter limits for the subranges.
Given in the following order: lower limit of 1, upper limit of 1 and lower limit of 2, upper limit of 2.
salsa_emission_mode#
- Fortran Type: C*20
-
Default: no_emission
Emission mode for aerosol and gaseous emissions.
Note that all chemical components included in the simulation must be activated via listspec. Also bc_aer_b = 'neumann' is required.
Currently 3 choices are available:
-
uniform
Sets a horizontally homogeneous surface flux of aerosols based on surface_aerosol_flux, aerosol_flux_dpg, aerosol_flux_sigmag, and aerosol_flux_mass_fracs_a.
-
parameterized
Sets surface fluxes based on the street type. The aerosol size distribution and mass composition of the emission is given similar to salsa_emission_mode = 'uniform'. The flux is then normalised based on the street type in the static input file and using emiss_factor_main, emiss_factor_side, main_street_id, side_street_id, and max_street_id.
-
read_from_file
Reads the emission information from the NetCDF aerosol-information file (see aerosol input file).
-
season_z01#
- Fortran Type: I
-
Default: 1
Modelling season.
Modelling season if the dry deposition parametrisation by Zhang et al. 2001 is applied via depo_pcm_par = 'zhang2001' and/or depo_surf_par = 'zhang2001'.
Currently 5 choices are available:
-
1
summer
-
2
autumn (no harvest yet)
-
3
late autumn (already frost)
-
4
winter
-
5
transitional spring
-
side_street_id#
- Fortran Type: I (99)
-
Default: 0
Index for identifying side streets following street type classes from
OpenStreetMap.Used if salsa_emission_mode = 'parameterized'.
sigmag#
- Fortran Type: R (7)
-
Default: [1.8, 2.16, 2.21, 2.0, 2.0, 2.0, 2.0]
The standard deviation of the log-normal aerosol number size distribution per aerosol mode.
A total of 7 different aerosol modes can be applied. Example aerosol modes are 'nucleation', 'Aitken', 'accumulation' and 'coarse'. In case of initializing_actions = 'set_constant_profiles', the initial aerosol size distribution is described by dpg, sigmag, and n_lognorm.
salsa_start_time#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: s
Time after which SALSA is switched on.
Can be used to allow the LES model to develop turbulence before aerosol particles and their processes are switched on.
surface_aerosol_flux#
- Fortran Type: R (7)
- Default: [104000000000.0, 32300000000.0, 5400000.0, 0.0, 0.0, 0.0, 0.0]
-
SI-Unit: m-2 s-1
The total surface aerosol number flux per aerosol mode.
A total of 7 different aerosol modes can be applied. Example aerosol modes are 'nucleation', 'Aitken', 'accumulation' and 'coarse'.
If salsa_emission_mode = 'uniform', the aerosol number emission is described by input parameters aerosol_flux_dpg, aerosol_flux_sigmag, aerosol_flux_mass_fracs_a, and surface_aerosol_flux.
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Enables/disables entire module.
When set to .T. will turn the entire module off without removing the namelist.
van_der_waals_coagc#
- Fortran Type: L
-
Default: .FALSE.
Üarameter to switch on the van der Waals forces when calculating the collision kernel in the coagulation subroutine.
Parametrisation follows Karl et al. (2016).
SLUrb Parameters#
aero_roughness_heat#
- Fortran Type: C*20
-
Default: kanda
Parametrization for the aerodynamic roughness lengths for heat for horizontal surfaces.
Parametrization used to compute the aerodynamic roughness lengths for heat (\(z_{0,h}\)) for horizontal surfaces.
Currently 2 choices are available:
-
kanda
Parametrization by Kanda et al. (2007) where \(z_{0,h}\) is dynamically computed at every time step.
-
fixed
\(z_{0,h}\) is fixed to a material default or user input value throughout the simulation.
-
anisotropic_street_canyons#
- Fortran Type: L
-
Default: .FALSE.
Enables anisotropic street canyons.
Enables anisotropic street canyons, i.e. separtion between walls and windows A and B. When false, average radiation for walls (windows) is used and prognostic equations are computed only for wall (window) A, with outputs of wall (window) B copied from A. The street canyon orientation can be set using either street_canyon_orientation in the namelist or
theta_canin netCDF input or both. If neither is set, an isotropic canyon is used in that grid cell regardless of value of this parameter. This allows mixing isotropic and anisotropic street canyons in the same simulation.
building_frontal_area_fraction#
- Fortran Type: R
-
Default: -9999.0
Frontal area fraction of buildings.
Ratio of building frontal area to total plan area (incl. natural surfaces/pervious surfaces).
building_height#
- Fortran Type: R
- Default: -9999.0
-
SI-Unit: m
Mean building height within the cell.
Mean building height within the cell. Note that a single-layer model like SLUrb does not perform very well with deep urban canopies (>60 m).
building_indoor_temperature#
- Fortran Type: R
- Default: 295.15
-
SI-Unit: K
Building indoor temperature.
Building indoor temperature. Kept constant throughout the simulation.
building_plan_area_fraction#
- Fortran Type: R
-
Default: -9999.0
Building plan area fraction.
Ratio of building plan area to total plan area incl. natural surfaces. Cannot exceed
urban_fraction.
building_type#
- Fortran Type: I
-
Default: 2
Building type.
Building type corresponding to building types as described in the SLUrb's model driver documentation. The building material properties are set according to the type.
deep_soil_temperature#
- Fortran Type: R
- Default: -9999.0
-
SI-Unit: K
Deep soil temperature.
Deep soil temperature to be used as a bottom boundary condition for the road model.
facade_resistance_parametrization#
- Fortran Type: C*20
-
Default: doe-2
Parametrization for the aerodynamic resistance between facade surfaces and the street canyon air.
Parametrization used to compute the aerodynamic roughness resistances for heat (\(r_{ah}\)) for facade surfaces (i.e. walls and windows).
Currently 3 choices are available:
-
doe-2
DOE-2 parametrization from the EnergyPlus building energy simulation system (EnergyPlus(TM) version 23.2.0 Engineering Reference, 2023), which is a function of natural convection strength, local surface roughness and street canyon effective wind speed. An average of windward and leeward parametrization are used.
-
krayenhoff&voogt
Parametrization following Krayenhoff & Voogt (2007), which is a function of local surface roughness and the street canyon effective wind speed.
-
rowley
Parametrization following Rowley (1932), which is a simple function of the street canyon effective wind speed.
-
moist_physics#
- Fortran Type: L
-
Default: .TRUE.
Enables moist dynamical processes in SLUrb.
Enables condensation/evaporation processes on roofs and roads as well as prognostic equation for street canyon mixing ratio. Only effective if humidity = .T., otherwise ignored.
n_layers_roads#
- Fortran Type: I
-
Default: 4
Number of road layers.
Number of road layers to be modelled. Non-default configuration requires setting of all road material parameters manually using the netCDF input. A minimum of three layers is required.
n_layers_roofs#
- Fortran Type: I
-
Default: 4
Number of roof layers.
Number of roof layers to be modelled. Non-default configuration requires setting of all roof material parameters manually using the netCDF input. A minimum of three layers is required.
n_layers_walls#
- Fortran Type: I
-
Default: 4
Number of wall layers.
Number of wall layers to be modelled. Non-default configuration requires setting of all wall material parameters manually using the netCDF input. A minimum of three layers is required.
n_layers_windows#
- Fortran Type: I
-
Default: 4
Number of window layers.
Number of window layers to be modelled. Non-default configuration requires setting of all window material parameters manually using the netCDF input. A minimum of three layers is required.
pavement_type#
- Fortran Type: I
-
Default: 2
Pavement type.
Pavement type corresponding to pavement types as described in the SLUrb's model driver documentation. The pavement material properties are set according to the type.
qsws_external#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: W m-2
Latent heat flux from external sources.
Additional latent heat flux from sources external to the model (e.g. industry or HVAC exhaust) per unit area. The flux is internally scaled to unit urban area and aggregated to atmospheric fluxes.
shf_external#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: W m-2
Sensible heat flux from external sources.
Additional sensible heat flux from sources external to the model (e.g. industry or HVAC exhaust) per unit area. The flux is internally scaled to unit urban area and aggregated to atmospheric fluxes.
shf_traffic#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: W m-2
Sensible heat flux from traffic.
Additional sensible heat flux from traffic per unit urban area. The flux is internally scaled and entered into the prognostic equation for street canyon air temperature.
street_canyon_aspect_ratio#
- Fortran Type: R
-
Default: -9999.0
Street canyon aspect ratio.
Street canyon aspect ratio (height-to-width ratio, \(H/W\)).
street_canyon_orientation#
- Fortran Type: R
- Default: -9999.0
-
SI-Unit: degrees
Street canyon orientation.
Only effective when anisotropic_street_canyons = .T. .
street_canyon_wspeed_factor#
- Fortran Type: C*20
-
Default: surfex
Factorfor the street canyon wind speed.
Parametrization for the factor used to compute street canyon wind speed from first atmospheric grid level wind speed.
Currently 3 choices are available:
-
surfex
The street canyon wind speed is computed as in the SURFEX model v8.1.
-
krayenhoff&voogt
The street canyon wind speed is computed following Krayenhoff and Voogt (2007).
-
masson
The street canyon wind speed is computed following Masson (2000).
-
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Disables the module.
Switch to turn off the module without having to remove slurb_parameters from the namelist.
urban_fraction#
- Fortran Type: R
-
Default: -9999.0
Urban fraction.
The plan area fraction of buildings and streets combined. The remaining surface area will be modelled as vegetation, water or open pavement surface by PALM-LSM depending on the user's setup. Any pervious or water surfaces should be included in the non-urban fraction, irregardless if they are part of urban fabric or not. As SLUrb's physical formulation is based on a street canyon, large paved areas without buildings (e.g. large parking lots) are better to be omitted from this fraction as well and modelled with PALM-LSM instead (surface type
pavement).
window_fraction#
- Fortran Type: R
-
Default: Depends on building_type
Window fraction.
Area fraction of windows of the total facade area. Setting the fraction to zero disables the window model.
Spectra Parameters#
averaging_interval_sp#
- Fortran Type: R
- Default: undefined
-
SI-Unit: s
Averaging interval for spectra output to local file DATA_1D_SP_NETCDF.
If this parameter has a non-zero value, temporally averaged spectra data are output. By default, spectra data are not subject to temporal averaging. The output interval length is limited by the parameter dt_dosp. In all cases, averaging_interval_sp <= dt_dosp must hold.
If an interval is defined, then by default the average is calculated from the data values of all time steps lying within this interval. The number of time steps entering into the average can be reduced with parameter dt_averaging_input_pr. If an averaging interval can not be completed at the end of a run, it will be finished at the beginning of the next restart run. Thus for restart runs, averaging intervals do not necessarily begin at the beginning of the run.
comp_spectra_level#
- Fortran Type: I (100)
-
Default: undefined
Vertical level for which horizontal spectra are to be calculated and output.
Levels are given as grid index
k. Spectra can be calculated for up to 100 vertical levels.
data_output_sp#
- Fortran Type: C*10 (10)
-
Default: undefined
Quantities for which horizontal spectra are to be calculated and output.
This is the general parameter to switch on calculation of spectra. Allowed standard quantities are data_output_sp = 'u', 'v', 'w', 'pt', 'q', 's'. Further quantities may be defined via the user-interface (see [../userint/output further quantities]).
Parameters comp_spectra_level and spectra_direction must be set too! Otherwise, no output will be generated. By default, spectra data are output in NetCDF format to the local file DATA_1D_SP_NETCDF. The temporal interval of the output times of spectra is assigned via the parameter dt_dosp. Spectra can also be temporally averaged (see averaging_interval_sp).
Spectra are calculated using the FFT-method defined by fft_method. Attention: Computation of spectra is currently not possible with fft_method = 'fftw'.
Calculation of spectra requires cyclic boundary conditions along the respective directions (see bc_lr and bc_ns).
dt_dosp#
- Fortran Type: R
- Default: undefined
-
SI-Unit: s
Temporal interval at which spectra shall be output.
Output can be skipped at the beginning of a simulation using parameter skip_time_dosp, which has zero value by default. Reference time is the beginning of the simulation, i.e. output takes place at times t = skip_time_dosp + dt_dosp, skip_time_dosp + 2 * dt_dosp, skip_time_dosp + 3 * dt_dosp, etc. The actual output times can slightly deviate from these theoretical values (see dt_dopr). If dt_dosp < dt, then spectra are output after each time step (it is advised to set dt_dosp = 0 if this kind of output is desired).
skip_time_dosp#
- Fortran Type: R
- Default: undefined
-
SI-Unit: s
Prohibits spectra data output until this interval has elapsed.
Counting from the beginning of the simulation t = 0 or dt_dosp.
Example:
If dt_dosp = 3600.0 and skip_time_dosp = 1800.0, then the first output will occur at t = 5400 s.
spectra_direction#
- Fortran Type: C*2 (10)
-
Default: undefined
Direction(s) along spectra are to be calculated.
For each quantity given by data_output_sp a corresponding direction must be assigned. E.g., if data_output_sp = 'u', 'w' is set, then spectra_direction = 'x', 'xy', means that spectra along
xwill be calculated foruand spectra alongxandywill be calculated forw. Note that spectra are always averaged perpendicular to their chosen direction, i.e. spectra alongxare calculated for each grid line along thex-direction and subsequently averaged alongy.Currently 3 choices are available:
-
x
Spectra are calculated for each grid line along the
x-direction and subsequently averaged alongy. -
y
Spectra are calculated for each grid line along the
y-direction and subsequently averaged alongx. -
xy
Spectra are calculated along the x- as well as along the y-direction and both spectra are output (no 2d-spectra calculation!).
-
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Switches off the module if set .T.
Synthetic Turbulence Generator Parameters#
boundary_layer_depth#
- Fortran Type: R
-
Default: 1000.0
Prescribed boundary-layer depth.
compute_velocity_seeds_local#
- Fortran Type: L
-
Default: .FALSE.
Switch to decide whether velocity seeds are computed locally or distributed over several processes.
If .F., the computation of the velocity seeds is done in parallel by multiple processes. This requires additional MPI communication to gather the calculated seeds on the respective processes responsible for the boundaries. This option is especially useful for large length scales where the computational effort can become huge (when computation of velocity seeds requires more time than MPI communication). If .T., velocity seeds are computed only locally and no MPI communication is necessary. This performs better when the turbulent length scales are small (when MPI communication requires more time than computation).
dt_stg_adjust#
- Fortran Type: R
- Default: 1800.0
-
SI-Unit: s
Time interval for adjusting turbulence statistics.
The turbulence statistics define the amplitude and sign of imposed correlations and are parametrized based on boundary-layer depth inferred from the inflow profiles as well as domain-averaged values of roughness, friction velocity and surface sensible heat flux. dt_stg_adjust only comes into effective if turbulence statistics are parametrized, i.e. if they are not read from the ASCII input file STG_PROFILES. dt_stg_adjust is mainly relevant in the case of a daily cycle, where the strength of turbulence, its integral length scales and boundary-layer depth change in time. Furthermore, dt_stg_adjust is also a useful parameter in case of more idealized setups with time-constant inflow profiles and desired parametrized turbulence statistics, i.e. the absence of the STG_PROFILES file. This case, dt_stg_adjust should be set to a small value in the order of a few hundreds of seconds. This is because at the beginning of the simulation domain averaged parameters are rapidly developing, meaning that the parametrized turbulence statistics change over time, too. If the turbulence statistics are regularly updated during this model spinup process, the imposed perturbations at the boundaries faster approach their final state compared to the case when dt_stg_adjust is set to a large value so that the imposed turbulence statistics are time-lagged to the actual domain-averaged surface conditions.
dt_stg_call#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: s
Time interval for calling synthetic turbulence generator.
Per default, the turbulence generator is called every time step (called every 0.0 seconds). Increasing dt_stg_call may help to reduce computational time.
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Switch to turn on/off the module.
If .T., the synthetic turbulence generator is not activated even if the namelist is present in the parameter file.
Surface Output Parameters#
averaging_interval_surf#
- Fortran Type: R
- Default: Value of averaging_interval
-
SI-Unit: s
Averaging interval for surface output data.
averaging_interval_surf <= dt_dosurf_av is required. If an interval is given, the average is calculated from all data of all time steps lying within this interval.
data_output_surf#
- Fortran Type: C*100 (300)
-
Default: undefined
List of surface quantities to be output.
Output of instantaneous data and temporally averaged data is possible. To output temporally averaged data, the name of the output quantity must be appended by the string '_av' (see data_output for more explanation). For example,
data_output_surf = 'shf', 'shf_av',will output instantaneous as well as time-averaged data of surface sensible heat flux 'shf' for all surface elements (surface grid points).
Depending on the specified output format (see to_netcdf and to_vtk), surface data for all surface elements is output to a netCDF file or/and into Fortran binary files.
Following quantities are available for output:
Quantity name Meaning SI-Unit albedo aggregated albedo value of the surface based on the tile albedo values - emissivity aggregated emissivity value for the surface based on the tile emissivity values - us friction velocity \(u_*\) m s-1 ts scaling parameter for temperature \(\vartheta_*\) K qs scaling parameter for mixing ratio \(q_*\) kg kg-1 ss scaling parameter for passive scalar - qcs scaling parameter for cloud water mixing ratio kg kg-1 ncs scaling parameter for number concentration of cloud water m-3 qrs scaling parameter for rain water mixing ratio kg kg-1 nrs scaling parameter for rain water droplet concentration m-3 ol Obukhov length m theta1 potential temperature at first grid point apart from (normal to) the surface K qv1 mixing ratio at first grid point apart (normal to) from the surface kg kg-1 thetav1 virtual potential temperature at first grid point apart from (normal to) the surface K usws surface momentum flux for the u-component (unit depending on flux_output_mode) m2 s-2 or N m-2 vsws surface momentum flux for the v-component (unit depending on flux_output_mode) m2 s-2 or N m-2 shf surface sensible heat flux (unit depending on flux_output_mode) K m s-1 or W m-2 qsws surface latent heat flux (unit depending on flux_output_mode) kg kg-1 m s-1 or W m-2 ssws surface passive scalar flux m s-1 qcsws surface flux of cloud water mixing ratio kg kg-1 m s-1 ncsws surface flux of number concentration of cloud water s-1 m-2 qrsws surface flux of rain water mixing ratio kg kg-1 m s-1 nrsws surface flux of rain water droplet concentration s-1 m-2 sasws surface salinity flux psu m s-1 q_surface surface moisture kg kg-1 theta_surface surface potential temperature K thetav_surface surface virtual potential temperature K theta_10cm 10-cm potential temperature K rad_net surface net radiation flux W m-2 rad_lw_in total incoming longwave radiation W m-2 rad_lw_dif incoming longwave radiation flux from sky only W m-2 rad_lw_out outgoing longwave radiation flux W m-2 rad_lw_ref incoming longwave radiation flux from reflections W m-2 rad_lw_res residual longwave radiation flux in surfaces after last reflection step W m-2 rad_sw_in total incoming shortwave radiation flux W m-2 rad_sw_dif incoming diffuse shortwave radiation flux W m-2 rad_sw_dir direct incoming shortwave radiation flux W m-2 rad_sw_out outgoing shortwave radiation flux W m-2 rad_sw_ref incoming shortwave radiation flux from reflections W m-2 rad_sw_res residual shortwave radiation flux in surfaces after last reflection step W m-2 rtm_skyvf sky view factor - rtm_skyvft sky view factor including transparancy of plant canopy - ghf ground heat flux W m-2 r_a aerodynamic resistance m-1 r_soil soil resistance m-1 r_canopy canopy resistance m-1 r_s total resistance m-1 uvw1 surface-parallel flow speed at first grid point apart from the surface m s-1 waste_heat waste_heat, only if indoor model is applied W m-2 im_hf heatflux at innermost building wall, only if indoor model is applied W m-2 z0 roughness length for momentum m z0h roughness length for heat m z0q roughness length for moisture m
dt_dosurf#
- Fortran Type: R
- Default: Value of dt_data_output
-
SI-Unit: s
Temporal interval at which instantaneous surface data shall be output.
dt_dosurf_av#
- Fortran Type: R
- Default: Value of dt_data_output_av
-
SI-Unit: s
Temporal interval at which time averaged surface data shall be output.
skip_time_dosurf#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: s
No surface data output before this interval has passed.
This parameter causes that surface data output starts only after this interval has passed (counting from the beginning of the simulation, t = 0).
skip_time_dosurf_av#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: s
No averaged surface data output before this interval has passed.
This parameter causes that averaged surface data output starts only after this interval has passed (counting from the beginning of the simulation, t = 0).
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Switch to turn on/off the module.
If switch_off_module = .T., output of surface data is not activated even if the namelist is present in the parameter file.
to_netcdf#
- Fortran Type: L
-
Default: .FALSE.
Control parameter to output surface data in netCDF format.
Surface data output in netCDF format requires parallel netCDF data format enabled with netcdf_data_format >= 5.
to_vtk#
- Fortran Type: L
-
Default: .FALSE.
Control parameter enabling surface data output to VTK format.
Surface data for each processor subdomain is output into a separate Fortran binary file. In a postprocessing step, these files are merged and converted into a single VTK file via surface_output_to_vtk, which can be used for visualization of surface quantities, e.g. by open source tool paraview.
Turbulent Inflow Parameters#
inflow_damping_height#
- Fortran Type: R
- Default: undefined
-
SI-Unit: m
Height below which the turbulence signal recycled.
In case of a turbulence recycling (see turbulent_inflow_method), this parameter defines the vertical thickness of the turbulent layer up to which the turbulence is extracted at the position of the recycling plane (see recycling_width), and then imposed to the inflow. Above this level, the turbulence signal is linearly damped to zero. The transition zone within which the signal falls to zero is controlled via parameter inflow_damping_width.
By default, inflow_damping_height is set as the height of the convective boundary layer as calculated from a precursor run. Information about proper settings for getting this CBL height from a precursor run can be found here.
inflow_damping_width#
- Fortran Type: R
- Default: Value of 0.1 * inflow_damping_height
-
SI-Unit: m
Transition range within which the turbulence signal is damped to zero.
For explanation see inflow_damping_height.
input_block_size#
- Fortran Type: I
-
Default: 30
Number of data input time layers read in per data input call.
In case of turbulent_inflow_method = 'read_from_file', data needs to be input from the dynamic input file. To avoid data input each timestep, boundary data is read in a blockwise manner, i.e. multiple time layers are read at once. With input_block_size the block size can be controlled by the user. The optimum block size is a trade-off between IO speed (usually the less IO calls the better) and memory consumption (too large block sizes could lead to interal memory faults). input_block_size must be >= 2 and must not exceed the subdomain size in x-direction.
recycling_width#
- Fortran Type: R
-
Default: undefined
Distance of the recycling plane from the inflow boundary (in m).
The parameter is only in effect when turbulence recycling (see turbulent_inflow_method) is used. This parameter sets the horizontal extension (along the direction of the main flow) of the so-called recycling domain which is used to generate a turbulent inflow. recycling_width must be larger than the grid spacing dx and smaller than the length of the total domain (nx * dx).
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Flag to switch on/off the user module.
If switch_off_module = .T., the user module is not activated even if the namelist is present in the parameter file.
turbulent_inflow_method#
- Fortran Type: C*80
-
Default: recycle_turbulent_fluctuation
Specifies the kind of the turbulent inflow method.
Different approaches to generate a turbulent inflow at the left boundary exit. There exist two general approaches, a recycling method and a read-from file method. With the recycling method, turbulent signals of all prognostic variables are recycled within a certain distance from the inflow boundary and are imposed onto the inflow boundary. With the read-from-file methode, inflow boundary data of the prognostic quantities are read from the dynamic input file and are imposed onto the inflow boundary.
A turbulent inflow requires Dirichlet conditions at the respective inflow boundary. So far, a turbulent inflow is realized from the left (west) side only, i.e. bc_lr = 'dirichlet/radiation' is required.
A few notes concerning the turbulence recycling: The distance of the recycling plane from the inflow boundary can be set with parameter recycling_width. The height above ground above which the turbulence signal is not used for recycling and the width of the layer within the magnitude of the turbulence signal is damped from 100% to 0% can be set with parameters inflow_damping_height and inflow_damping_width.
For the recycling method, the initial (quasi-stationary) turbulence field must be generated by a precursor run and used via setting initializing_actions = 'cyclic_fill'. This is not necessarily required if turbulent_inflow_method = 'read_from_file' is set, where also initializing_actions = 'set_constant_profiles' or *'read_from_file' is an option.
A detailed setup for a turbulent inflow using the turbulence recycling method is described in here.
Currently 4 choices are available:
-
read_from_file
Turbulent inflow is realized using a direct forcing method, where yz-cross-sections of the prognostic variables are read from the dynamic input file and are imposed onto the inflow boundary with high temporal resolution. The respective cross-sections can be obtained from a precursor run. The pre-processing tool palm_cti (link!) processes the yz-cross-section data accordingly and write it into the dynamic input file.
The boundary data must be generated by a precursor simulation which features the same grid spacing along the x, y, z-dimension as well as the same domain height. The domain extent along the y dimension of the precursor simulation can be smaller or equal the domain extent along y of the main simulation. Starting from the point in time where the precursor simulation reaches a quasi-stationary state (yz cross-section output can be skipped via skip_time_do2d_yz), it is recommended to output yz-cross-section data each timestep, in order to provide high temporal resolution inflow data. The provided output interval of the precursor simulation needs to cover the entire simulation time of the main simulation.
It is recommended to set initializing_actions = 'cyclic_fill', though turbulent_inflow_method = 'read_from_file' also works with other initialization methods.
Compared to the turbulent recycling method, this direct method is more robust in setups where the boundary-layer characteristics change. Even complete daily cycles can be considered.
-
recycle_turbulent_fluctuation
Turbulent fluctuations of all prognostic variables are recycled and added to the mean inflow profile. A turbulent inflow is realized using the turbulence recycling method from Lund et al. (1998, J. Comp. Phys., 140, 233-258) modified by Kataoka and Mizuno (2002, Wind and Structures, 5, 379-392).
Attention'
If surface heating/cooling or a surface waterflux is applied, a horizontal temperature (humidity) gradient inside the boundary layer will build up, because the temperature/humidity profiles at the inflow are constant in time. The resulting horizontal differences in buoyancy can trigger an undesired circulation inside the entire domain and instabilities at the inflow boundary (see pt_damping_factor). -
recycle_absolute_value
This is a variation of the original recycling method, where the absolute instantaneous values of all prognostic variables are recycled, so that the values at the inflow boundary and the recycling plane are identical (for vertical velocity, still only the turbulent fluctuations are recycled). With this method there is no horizontal temperature (humidity) gradient and thus any thermal circulation and instabilities at the inflow boundary will not occur. Note that the mean inflow profiles of all prognostic variables but vertical velocity will now change in time, which means that the boundary layer height at the inflow is changing.
-
recycle_absolute_value_thermodynamic
This is a further variation of the original recycling method, where the absolute instantaneous values of potential temperature and (if present) humidity are recycled. For all other prognostic variables only the turbulent fluctuations are recycled. With this method there is no horizontal temperature (humidity) gradient and thus any thermal circulation and instabilities at the inflow boundary will not occur. Note that the mean inflow profiles of potential temperature (humidity) will now change in time (i.e. the boundary layer height at the inflow is changing), in contrast to the inflow profile of all other quantities (e.g. u, v, w) that are constant. In order to avoid this mismatch, the boundary layer height should and can be kept constant by applying a large_scale_subsidence to scalar quantities.
-
Urban Surface Parameters#
building_type#
- Fortran Type: I
-
Default: 1
Building type according to internal classification.
Building type according to internal classification my means of the year of construction and usage. This type is assumed for all buildings that are present in the simulation setup. Individual building types can be provided via static driver file (see [building_type(#urban_surface_parameters--building_type) = 0.
At the moment six types of buildings are offered as outlined in the following table:
building_type Description albedo_type Notes 0 User defined - All building parameters must be explicity set by the user in the static driver file 1 Residential building, built before 1950 33 2 Residential building, built from 1950-2000 33 3 Residential building, built after 2000 33 4 Non-residential building, built before 1950 33 5 Non-residential building, built from 1950-2000 33 6 Non-residential building, built after 2000 33
roof_category#
- Fortran Type: I
-
Default: 2
Roof category.
roof_inner_temperature#
- Fortran Type: R
- Default: 295.0
-
SI-Unit: K
Initial temperature of the inner roof surface (inside the buildings).
This parameter is only used, if the indoor model is not used.
roughness_concrete#
- Fortran Type: R
- Default: 0.001
-
SI-Unit: m
Average roughness length of concrete surfaces.
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
If [switch_off_module]((#urban_surface_parameters--switch_off_module) = .T., the urban surface model is switched off, despite the namelist is set.
wall_category#
- Fortran Type: I
-
Default: 2
Wall category for wall surface over pedestrian zone.
wall_inner_temperature#
- Fortran Type: R
- Default: 295.0
-
SI-Unit: K
Initial temperature of the inner wall surface (inside the buildings).
This parameter is only used, if the indoor model is not used.
window_inner_temperature#
- Fortran Type: R
- Default: 295.0
-
SI-Unit: K
Initial temperatur of the inner window surface (inside the buildings).
This parameter is only used, if the indoor model is not used.
User Parameters#
data_output_masks_user#
- Fortran Type: C*30 (300,100)
-
Default: undefined
User-defined quantities for which masked data are to be output (see masked data output).
Beside the standard output quantities (which can be selected via parameter data_output_masks), the user can define (without any restriction) additional output quantities (e.g. the temperature variance, etc.) of masked data. Each of these quantities must be given a unique identifier (string), which must be different from the strings that define the standard output quantities (see list in the description of data_output_masks). Data output can be switched on by assigning the respective strings to data_output_masks_user.
The user must calculate/provide the respective data array(s) by appropriately extending the user interface.
For further steering of masked output (output times, defining mask locations, time averaging, etc.), the steering parameters for data_output_masks apply accordingly.
The first dimension of the shape depends on the variable
max_masksin source file modules.f90.
data_output_pr_user#
- Fortran Type: C*30 (300)
-
Default: undefined
User defined quantities for which horizontally averaged profile data is to be output.
Beside the standard output quantities (which can be selected via parameter data_output_pr), the user can define (without any restriction) additional output quantities (e.g. turbulent resolved-scale horizontal momentum fluxes, etc.). Each of these quantities must be given a unique identifier (string), which must be different from the strings that define the standard output quantities (see list in the description of data_output_pr). Data output can be switched on by assigning the respective strings to data_output_user.
The user must calculate/provide the respective data array(s) by appropriately extending the user interface.
data_output_user#
- Fortran Type: C*30 (500)
-
Default: undefined
User defined quantities for which 2d cross section and/or 3d volume data are to be output.
Beside the standard output quantities (which can be selected via parameter data_output), the user can define (without any restriction) additional output quantities (e.g. the temperature variance, etc.). Each of these quantities must be given a unique identifier (string), which must be different from the strings that define the standard output quantities (see list in the description of data_output). Data output can be switched on by assigning the respective strings to data_output_user.
The user must calculate/provide the respective data array(s) by appropriately extending the user interface.
For further steering of the user defined output (output times, cross section levels, time averaging, etc.), the steering parameters for data_output apply accordingly.
Example:
If the user defines a new output quantity named 'pt2', then output of horizontal cross sections can be selected by assigning data_output_user = 'pt2_xy'.
region#
- Fortran Type: C*40 (9)
-
Default: total domain
Names of the subdomains defined by the user.
With this variable, names can be assigned to the subdomains defined by the user (see statistic_regions). Up to 9 subdomains are allowed (region(1) to region(9)). The total domain has the index 0 (region(0)).
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Flag to switch on/off the user module.
If switch_off_module = .T., the user module is not activated even if the namelist is present in the parameter file.
UV Radiation Parameters#
num_reflections#
- Fortran Type: I
-
Default: 0
Number of reflection steps for UV radiation to be performed inside the RTM.
The number of reflection steps for the reflected UV radiation between mutually visible surfaces. Since UV radiation is spectrally resolved, each wavelength is treated separately. Reflections are treated identical as in the radiative transfer model, where reflections are considered as purely Lambertian (diffuse), i.e. no specular reflections are considered. The corresponding surface-type dependent albedo is taken from pre-defined bulk parameters.
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Switch to turn off the module.
If switch_off_module = .T., the UV radiation model is not activated even if the namelist is present in the parameter file.
uv_integration_method#
- Fortran Type: C*50
-
Default: from_irradiance
Methods used to compute the UV irradiance.
This parameter describes the method used to compute UV-radiances at the surface including partial shading and reflections. Values may be combined, separated by one or more blanks, i.e. uv_integration_method = 'from_irradiance from_radiance'. The usage of this UV module requires the RTM switched-on with radiation_interactions_on to compute realistic shading and reflections. The UV irradiance is computed from externally provided UV irradiances considering the obstruction of the upper hemisphere by buildings, tall canopy and other transparent or non-transparent obstacles. Therefore, for the actual sun zenith and azimuth angle, the corresponding UV scenario is read from an external file PIDS_UV, preprocessed by the tool uv2palm, which computes spectrally-resolved UV irradiances and UV radiances using the LIBRADTRAN library (Mayer and Kylling, 2005). The spectral resolution as well as the number of considered sun-zenith angles can be freely chosen with uv2palm. The UV model is purely diagnostic and independent on the computed flow, i.e. if only the UV radiance is of interest, it is sufficient to run a spinup only (see spinup_time), and to enable output via data_output_during_spinup. The UV model is called each 2d horizontal cross section output step as given by dt_do2d_xy.
Currently 2 choices are available:
-
from_irradiance
The UV irradiance is computed from externally-provided spectrally resolved UV irradiances separated into a direct and diffuse portion. The external values are multiplied with the local sky-view factor and transparency inferred from the radiative transfer model. This method neglects the effect of non-isotropic diffuse UV radiation on shading patterns.
-
from_radiance
The UV irradiance is computed from externally-provided spectrally resolved UV radiances. The discrete resolution along azimuth and zenith can be freely chosen with uv2palm, though the spherical resolution of the UV input file needs to correspond to the settings of raytrace_discrete_azims and raytrace_discrete_elevs. With this approach, the spherical directions obstructed by transparent or non-transparent obstacles are stored during the initialization. The local UV irradiance is then computed by integration over all azimuth and zenith angles neglecting spherical directions obstructed by non-transparent obstacles. Spherical directions obstructed by transparent obstacles are considered, but the radiance originating from these directions is reduced by the reflected and absorbed portions at the obstacles, i.e. only the transmitted portion is considered. With this method, the effect of non-isotropic diffuse radiation is considered.
-
Virtual Flight Parameters#
flight_angle#
- Fortran Type: R (500)
- Default: 45.0
-
SI-Unit: degree
Horizontal flight angle describing the flight direction.
The parameter only has an effect if leg_mode = 'cyclic'. Else, the flight direction is determined by x_start, x_end, y_start and y_end.
The flight_angle must be within 0 and 360 degrees. The convention is as following:
- flight_angle = 0.0 - aircraft moves to the east
- flight_angle = 90.0 - aircraft moves to the north
- flight_angle = 180.0 - aircraft moves to the west
- flight_angle = 270.0 - aircraft moves to the south

flight_begin#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: s
Start time of virtual flight measurement.
Relative time since start of the 3D-model.
flight_end#
- Fortran Type: R
- Default: undefined
-
SI-Unit: s
End time of virtual flight measurement.
Relative time since start of the 3D-model. By default, the flight is continued until the end of the simulation.
flight_level#
- Fortran Type: R (500)
- Default: 100.0
-
SI-Unit: m
Flight level.
Flight level relates to the bottom model boundary at
z = 0.
leg_mode#
- Fortran Type: C*6 (500)
-
Default: cyclic
Flag to choose steering of flight mode.
Currently 2 choices are available:
-
cyclic
Starting at (x_start, y_start), the flight direction is determined only by flight_angle and the sensor moves cyclically through the model domain. Parameters x_end and y_end are ignored.
-
return
The flight direction is determined by (x_start, y_start) and (x_end, y_end). The sensor turns around at the end-position and flies back, where it turns around again at the start-position and so on, until flight_end is reached. Parameter flight_angle is ignored.
-
max_elev_change#
- Fortran Type: R (500)
-
Default: 0.0
Maximum possible elevation change if rate_of_climb \(\neq\) 0.0.
If the maximum elevation change is reached, the virtual aircraft starts descending/ascending until the original start level is reached again. For longer flights, the sensor moves vertically between flight_level and flight_level + max_elev_change or flight_level - max_elev_change depending on the sign of rate_of_climb.
Please note, flight_level +/- max_elev_change must not exceed the top/bottom of the model domain.
rate_of_climb#
- Fortran Type: R (500)
- Default: 0.0
-
SI-Unit: m s-1
Vertical speed of the aircraft.
Positive and negative values are allowed.
speed_agl#
- Fortran Type: R (500)
- Default: 25.0
-
SI-Unit: m s-1
Ground speed of the aircraft.
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Flag to switch on/off the module.
If switch_off_module = .T., the virtual flight module is not activated even if the namelist is present in the parameter file.
x_end#
- Fortran Type: R (500)
- Default: undefined
-
SI-Unit: m
End-position of the flight leg along
x.This parameter is mandatory if leg_mode = 'return'. If leg_mode = 'cyclic', the parameter is ignored.
x_start#
- Fortran Type: R (500)
- Default: undefined
-
SI-Unit: m
Start-position of the flight leg along
x.This parameter is mandatory.
y_end#
- Fortran Type: R (500)
- Default: undefined
-
SI-Unit: m
End-position of the flight leg along
y.This parameter is mandatory if leg_mode = 'return'. If leg_mode = 'cyclic', the parameter is ignored.
y_start#
- Fortran Type: R (500)
- Default: undefined
-
SI-Unit: m
Start-position of the flight leg along
y.This parameter is mandatory.
Virtual Measurement Parameters#
dt_virtual_measurement#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: s
Temporal interval at which virtual measurements shall be output.
It is obligatory to set this parameter. It must not be zero. Therefore, the default value will cause the run to terminate.
dt_virtual_measurement_pr#
- Fortran Type: R
- Default: Value of (dt_virtual_measurement)
-
SI-Unit: s
Temporal interval at which virtual profile measurements shall be output.
Must not be zero.
dt_virtual_measurement_tr#
- Fortran Type: R
- Default: Value of (dt_virtual_measurement)
-
SI-Unit: s
Temporal interval at which virtual trajectory measurements shall be output.
Must not be zero.
dt_virtual_measurement_ts#
- Fortran Type: R
- Default: Value of (dt_virtual_measurement)
-
SI-Unit: s
Temporal interval at which virtual time series measurements shall be output.
Must not be zero.
interpolate_to_location#
- Fortran Type: L
-
Default: .FALSE.
Flag to switch on/off interpolation onto the exact sampling coordinates.
If interpolate_to_location = .T., the output variables will be bi-linearly interpolated onto the given sampling location, else, a simple nearest neighbor approach is used to sample the variables on the numeric grid. Please note, this feature is only implemented for single-point measurements at the moment, not for trajectory, profile, or multi-point timeseries measurements where off_ts > 0 or off_ts_z > 0. Furthermore, only atmospheric variables are considered for interpolation, surface or soil variables are not be interpolated and always refer to the grid center.
If interpolate_to_location = .F., all sampled quantities refer to the grid center, also the wind-velocity components, which are linearly interpolated from the staggered grid onto the grid center in this case. To determine the respective grid point, a simple nearest neighbor method is employed.
off_pr#
- Fortran Type: I
-
Default: 0
Horizontal sampling radius for profile measurements.
The sampling radius is given in grid points horizontally placed around the original measurement location (for profile measurements). The sampling set includes the grid-normal and diagonal grid points. As an example, off_pr = 1 will output 9 profiles (3x3 matrix with the original sampling location in the middle).
Note:
The value of off_pr must not be larger than the number of ghost points minus one, so for the Wicker-Skamarock advection scheme (see momentum_advec) 0 <= off_pr <= 2 must hold. The Piascek-Willimas advection scheme only allows off_pr = 0.
off_pr_z#
- Fortran Type: I
-
Default: 0
Vertical sampling radius for profile measurements.
Number of grid points along
zaround the original measurement location (for profile measurements).
off_tr#
- Fortran Type: I
-
Default: 0
Horizontal sampling radius for trajectory measurements.
The sampling radius is given in grid points horizontally placed around the original measurement location (for trajectory measurements). The sampling set includes the grid-normal and diagonal grid points. As an example, for a grid point on the trajectory, off_tr = 1 will output the original grid point + the 8 horizontally surrounding grid points. Note:
The value of off_tr must not be larger than the number of ghost points minus one, so for the Wicker-Skamarock advection scheme (see momentum_advec) 0 <= off_tr <= 2 must hold. The Piascek-Willimas advection scheme only allows off_tr = 0.
off_tr_z#
- Fortran Type: I
-
Default: 0
Vertical sampling height for trajectory measurements.
Number of grid points along
zaround the original measurement location (for trajectory measurements).
off_ts#
- Fortran Type: I
-
Default: 0
Horizontal sampling radius for timeseries measurements.
The sampling radius is given in grid points horizontally placed around the original measurement location (for timeseries measurements). The sampling set includes the grid-normal and diagonal grid points. As an example, off_ts = 1 will output 9 timeseries (3x3 matrix with the original sampling location in the middle).
Note:
The value of off_ts must not be larger than the number of ghost points minus one, so for the Wicker-Skamarock advection scheme (see momentum_advec) 0 <= off_ts <= 2 must hold. The Piascek-Willimas advection scheme only allows off_ts = 0.
off_ts_z#
- Fortran Type: I
-
Default: 0
Vertical sampling height for timeseries measurements.
Number of grid points along
zaround the original measurement location (for time series measurements).
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Flag to switch on/off module.
If switch_off_module = .T., the virtual measurement module is not activated even if the namelist is present in the parameter file.
vm_time_start#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: s
Start time of the virtual measurements.
vm_time_start refers to the simulation start. Sampling will start at origin_date_time + vm_time_start. Time information given in the virtual-measurement driver file is ignored at the moment.
warn_out_of_domain_locations#
- Fortran Type: L
-
Default: .FALSE.
Flag to switch on/off warnings for timeseries sampling.
If warn_out_of_domain_locations = .T., warnings will be given when the given sampling location is not within the model domain. Please note, warnings will be only given for point measurements, not for trajectory or profile measurements.
Wind Turbine Parameters#
air_density#
- Fortran Type: R
- Default: 1.225
-
SI-Unit: kg m-3
Air density used for turbine torque and power calculation.
dt_data_output_wtm#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: s
Output interval for NetCDF data.
Output occurs at the next closest time step after each integer multiple of dt_data_output_wtm. With the default value \(0.0\), data will be output every time step.
gear_efficiency#
- Fortran Type: R
-
Default: 1.0
Efficiency of the gear.
Default value means an efficiency of 100%.
gear_ratio#
- Fortran Type: R
-
Default: 97.0
Gear ratio from rotor to generator.
generator_efficiency#
- Fortran Type: R
-
Default: 0.944
Electric efficiency of the generator.
generator_inertia#
- Fortran Type: R
- Default: 534.116
-
SI-Unit: kg m2
Moment of inertia of the generator.
generator_power_rated#
- Fortran Type: R
- Default: 5296610.0
-
SI-Unit: W
Rated mechanical (rotor) power of the wind turbine.
generator_speed_rated#
- Fortran Type: R
- Default: 121.6805
-
SI-Unit: rad s-1
Rated generator speed.
generator_torque_max#
- Fortran Type: R
- Default: 47402.91
-
SI-Unit: N m
Maximum Torque of the generator.
Should be set to a value 10% above rated.
generator_torque_rate_max#
- Fortran Type: R
- Default: 15000.0
-
SI-Unit: N m s-1
Maximum generator torque increase rate.
hub_x#
- Fortran Type: R (10000)
- Default: undefined
-
SI-Unit: m
x-coordinate of the hub.
If n_turbines > 1, values need to be given for each turbine separately. Example for three turbines:
hub_x = 1000.0, 1500.0, 2000.0,Note that for turbines which are placed into child domains, the x-coordinate refers to the origin of the child, which is 0.0 at its left boundary.
hub_y#
- Fortran Type: R (10000)
- Default: undefined
-
SI-Unit: m
y-coordinate of the hub.
If n_turbines > 1, values need to be given for each turbine separately. Example for three turbines:
hub_y = 1000.0, 1500.0, 2000.0,Note that for turbines which are placed into child domains, the y-coordinate refers to the origin of the child, which is 0.0 at its left boundary.
hub_z#
- Fortran Type: R (10000)
- Default: undefined
-
SI-Unit: m
z-coordinate of the hub (hub height).
If n_turbines > 1, values need to be given for each turbine separately. Example for three turbines:
hub_z = 150.0, 150.0, 150.0,Note that for turbines which are placed into vertically shifted child domains (see domain_layouts), the z-coordinate refers to the origin of the root domain, which is 0.0 at its bottom.
n_airfoils#
- Fortran Type: I
-
Default: 8
Number of airfoils used in the turbine model.
The airfoil data should be provided in the additional input file
<run_identifier>_wtm(see here for a file describing the NREL 5 MW turbine).
n_turbines#
- Fortran Type: I
-
Default: 1
Number of wind turbines.
n_turbines <= 10000 is required.
nacelle_cd#
- Fortran Type: R (10000)
-
Default: 0.85
Drag coefficient of the nacelle.
nacelle_cd and nacelle_radius both must be non-zero if the nacelle should be included. If n_turbines > 1, values need to be given for each turbine separately.
nacelle_radius#
- Fortran Type: R (10000)
- Default: 0.0
-
SI-Unit: m
Radius of the nacelle of each turbine.
nacelle_radius and nacelle_cd both must be non-zero if the nacelle should be included. A nacelle with a spherical shape and a drag coefficient nacelle_cd is assumed. If n_turbines > 1, values need to be given for each turbine separately.
pitch_angle#
- Fortran Type: R (10000)
- Default: 0.0
-
SI-Unit: degree
Constant pitch angle of the blades that is used if pitch_control = .F..
If n_turbines > 1, values need to be given for each turbine separately.
pitch_control#
- Fortran Type: L
-
Default: .FALSE.
Parameter to switch on the pitch control.
This parameter controls the pitch angle such that constant power is produced for wind speeds greater than the rated wind speed. The parameter generator_power_rated is used here.
pitch_rate#
- Fortran Type: R
- Default: 8.0
-
SI-Unit: degree s-1
Maximum pitch rate.
region_15_min#
- Fortran Type: R
- Default: 70.16224
-
SI-Unit: rad s-1
Lower generator speed boundary of region 1.5.
Region 1.5 is a start-up region between regions 1 and 2 that represents the lower limit of the generator speed, which limits the operational speed range of the wind turbine. Note, region 1 is before cut-in wind speed. Here, wind is used to accelerate the rotor so no power is produced. See Jonkman et al., 2009 for more details.
region_2_min#
- Fortran Type: R
- Default: 91.21091
-
SI-Unit: rad s-1
Lower generator speed boundary of region 2.
region_2_slope#
- Fortran Type: R
- Default: 2.332287
-
SI-Unit: N m (rad s-1)-2
Slope constant for region 2.
In region 2, a constant tip speed ratio is maintained to optimize the power capture. In this region, the generator torque is proportional to the square of the filtered generator speed. See Jonkman et al., 2009 for more details.
rotor_inertia#
- Fortran Type: R
- Default: 34784179.0
-
SI-Unit: kg m2
Moment of inertia of the rotor.
rotor_radius#
- Fortran Type: R (10000)
- Default: 63.0
-
SI-Unit: m
Radius of the rotor.
If n_turbines > 1, values need to be given for each turbine separately.
rotor_speed#
- Fortran Type: R (10000)
- Default: 0.9
-
SI-Unit: rad s-1
Initial rotational speed for each turbine.
If n_turbines > 1, values need to be given for each turbine separately.
segment_length_tangential#
- Fortran Type: R
-
Default: 1.0
Tangential length of rotor segments.
The segment length is given as factor of the minimum grid spacing MIN(dx,dy,dz).
segment_width_radial#
- Fortran Type: R
-
Default: 0.5
Radial width of rotor segments.
The segment width is given as factor of the minimum grid spacing MIN(dx,dy,dz).
smearing_kernel_size#
- Fortran Type: R
-
Default: 2.0
Size of the smearing kernel given as multiples of the grid spacing dx.
For details, refer to https://palm.muk.uni-hannover.de/trac/wiki/doc/tec/wtm. A large smearing kernel results in an overestimation of tip speed ratio and power output. For coarse grid spacings (e.g., 12 grid points per rotor diameter) the power overestimation can be as large as 25%. This error can be reduced by setting smearing_kernel_size = 1.0. Do not use values below 1.0 because then the number of grid cells inside the smearing kernel becomes very small (<10). In this case the sum of the body forces acting on the flow may be lower than the force acting on the rotor blade.
speed_control#
- Fortran Type: L
-
Default: .FALSE.
Parameter to switch on the speed control.
This parameter controls the rotational speed of the turbine for wind speeds less than rated wind speed.
switch_off_module#
- Fortran Type: L
-
Default: .FALSE.
Parameter to switch off the entire wind turbine module.
switch_off_module = .T. can be used to switch off this module without removing (or commenting) the namelist from the namelist file.
tilt_angle#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: degree
Tilt angle of the rotor plane.
A positive angle means the bottom of the rotor plane is tilted away from the tower to avoid the blades hitting the tower.
time_turbine_on#
- Fortran Type: R
- Default: 0.0
-
SI-Unit: s
Simulation time at which the turbines are switched on.
tip_loss_correction#
- Fortran Type: L
-
Default: .FALSE.
Parameter to switch on the tip loss correction.
tower_cd#
- Fortran Type: R (10000)
-
Default: 1.2
Drag coefficient of the tower.
If n_turbines > 1, values need to be given for each turbine separately.
tower_diameter#
- Fortran Type: R (10000)
- Default: 0.0
-
SI-Unit: m
Diameter of the cylindrical tower of the wind turbine.
This parameter must be non-zero if a tower should be included. The parameter must be specified for every turbine separately.
yaw_angle#
- Fortran Type: R (10000)
- Default: 0.0
-
SI-Unit: degree
Yaw angle for each turbine.
A positive yaw means a rotation of the rotor in clockwise direction within the xy-plane. At a yaw angle of zero degrees the rotor is facing west.
yaw_control#
- Fortran Type: L
-
Default: .FALSE.
Parameter to switch on the yaw controller.
If yaw_control = .F., then the constant yaw angle specified by yaw_angle will be used. If set yaw_control = .T., then the yaw angle will adjust according to the local wind direction, as soon as the yaw misalignment reaches yaw_misalignment_max until yaw_misalignment_min is reached. The rotational speed of the yaw mechanism is controlled by yaw_speed.
yaw_misalignment_max#
- Fortran Type: R
- Default: 0.08726
-
SI-Unit: rad
Maximum tolerated yaw misalignment.
yaw_misalignment_min#
- Fortran Type: R
- Default: 0.008726
-
SI-Unit: rad
Minimum yaw misalignment for which the actuator stops.
yaw_speed#
- Fortran Type: R
- Default: 0.005236
-
SI-Unit: rad s-1
Speed of the yaw actuator.